缩醛基的形成和水解
我们已经描述了一些实验结果,它们说明了立体电子效应对缩醛基的构型和构象的影响.接着的一个相关问题是,这些立体电子效应在这个基团的形成和水解过程中是否起着类似的作用.
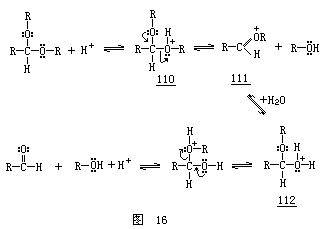 缩醛基的形成或水解是通过图
16
中所描述的机制进行的,其中氧鎓离子和半缩醛是中间体.缩醛水解过程中的决速步也已被
缩醛基的形成或水解是通过图
16
中所描述的机制进行的,其中氧鎓离子和半缩醛是中间体.缩醛水解过程中的决速步也已被
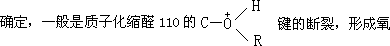
氧 3 鎓离子 111.而后,这个离子迅速水合,产生质子化的半缩醛 112,112 在适当的质子转移之后,可以给出醛产物.因而,弄清立体电子效应是否影响这一水解反应的决速步(110→111)是至关紧要的.
我们首先考察,当考虑立体电子效应时,将可能发生什么情况.二甲氧基甲烷的构象 D、A 和 C 分别有两个、一个和零个顶端异构效应(图 17),对这些构象的分析导致了下面的预测.构象体 A 中的C—OR 键应该比正常的C— O 醚键短,因为顶端异构效应使它具有部分的双键特征;而 C—*OR 键应该比通常的长,这起因于另一个氧的电子给予.在构象体 D 中,两个 C—O 键都具有部分的双键特征,它们可能比正常的短.既然构象体 C 没有顶端异构效应, 它的两个氧是等同的;C—O 键的长度应该是正常的,其数值介于构象体 A 的预测值之间.
从甲基吡喃糖甙和其它产品的 X 射线晶体结构测定的实验数据中采集的键长数据强有力地支持上面的讨论.带有一电子对与 C—*OR 键处于反式共平面的氧原子一定有一个较短的 C—O 键,而 C—*OR 键要比正常的长.更有意义的是,理论计算的结果与上面的讨论和实验观察相符合.
因此立体电子效应也应该影响缩醛基的氧的碱性,由此而影响它们质子化的相对容易程度.例如在构象体 A 中,O*—R 基团的氧原子的碱性应该比另一个氧原子的强.在没有顶端异构效应的构象体 C 中,两个氧原子是完全等同的,它们所具有的碱性应该介于构象体 A 的氧原子的碱性之间.由于构象体 D有两个顶端异构效应,两个氧的碱性都应该比构象体 A 的 OR 氧的碱性略强.结果,通过 O*R 氧的质子化,构象体 A 中(→113)C—O*R 键长增长的程
 子对与度应该比构象体
C 中(→114)的大,因为 113 的非质子化的氧有一电
子对与度应该比构象体
C 中(→114)的大,因为 113 的非质子化的氧有一电
处于反式共平面,而 114 的情况不同.对于构象体 D,O*R
氧的电子对与 C—OR 键处于反式共平面,它的质子化(→115)应该导致一种和 113
对上→
相类似的情况.在另一方面,O*R 氧的质子化可以发生在另一个电子
116),在这种情形中, 键长不应该增加如 115 那么多,
因为在116 中,质子化的氧仍有一电子对与 C—OR 键处于反式共平面.这样在构象体D 中,O*R 氧的两个电子对之一的立体特异的质子化导致了两种完全不同的情况(115 和 116).这是非常有趣的,因为它指出了质子化立体化学的重要性以及同一个氧原子上两个电子对的相对碱性.例如在构象体 D 中,O*R 氧在顶端异构效应中没有被占用的电子对应该比另一对的碱性强,据此,D 的质子化应该生成比 115 占优势的 116.
 对质子化的二羟基甲烷的四种不同构象已经做了从头算起的研究.把这些结果和从相应的中性构象所得到的结果进行比较表明,键长和重叠比例显示了很强的立体电子的构象依赖性,这与上面的讨论完全一致.
对质子化的二羟基甲烷的四种不同构象已经做了从头算起的研究.把这些结果和从相应的中性构象所得到的结果进行比较表明,键长和重叠比例显示了很强的立体电子的构象依赖性,这与上面的讨论完全一致.
键
现在,我们可以考察质子化形式 113、114、115 和 116 中的的断裂.在相邻氧原子的一电子对的帮助下,中间体 113 和 115
可以断裂给出氧鎓离子 117 和醇.在 116 中发生类似的情况,但 RO*H 的离去应该比较困难, 因为离去基团的氧原子仍有一个顶端异构效应.构象体 114 不能在一电子对的帮助下发生断裂直接给出离域的离子 117,它应该首先生成高能量非离域的正离子 118,在 C-OR 键旋转 60°之后,118 就能够转变成较稳定的氧鎓离子 117.如果 114→118→117 过程与 115→117 或 113→117 过程之间的能量差达到前者不能与后二者相竞争时,则此缩醛水解反应即可认为是在立体电子控制下发生的.
由于立体电子控制原理,其逆过程,即醇对氧鎓离子的加成过程(如 117
→113),一定是通过一严格限制的途径发生的.有关这一途径的证据来自于含有能紧密地相互趋近的一个胺基和一个羰基的分子的 x 射线分析(如 119)
(亦参考文献[82]).
Bürgi、Dunitz 和 Shefter 作了如下观察:发现 N:⋯C=O 距离作为一个键太长了,但作为一个非键却短得太多, RRC=O 单元(120→121)偏离了它通常的共平面形状,并且假设趋近的氮原子的电子对的可能取向(这不能观察到)位于靠近三级胺基的三重轴上.这一分析清楚地表明了氮原子和羰基之间的相互作用.晶体结构毫无疑问地证实,亲核试剂沿着一条与 C=O 键夹角接近于 109°的路线趋近羰基.当亲核试剂接近羰基碳时,氧原子和烷基取代基向外弯曲而偏离平面,而且碳氧键长增长(见 120→121).
已经进行过氢负离子对甲醛进行亲核加成而产生甲氧负离子(H-+C=O
→CH3O-)的 SGF-LCGO 计算.有趣的是,对于这个反应,计算出的反应途径表
明,它与从胺对酮基加成的结构相关性得出的计算结果具有惊人的相似性. 亲核试剂对羰基加成反应路径的强有力证据,来自于萘胺基酮 122 的 X
射线结构.在多数 1,8 二取代的萘中,两个取代基都向外伸展(见 123).在122 中,C-CO 键是向外伸展的,但 C-N 键却向内倾斜(见 124).和在未扭曲的分子中相比,这使氮处在较有力的进攻位置. 从胺基酯( 122 , COCH3=COOCH3)和胺基羧酸(122,COCH3=COOH)观察到了类似的情况.
Capon 和 Thacker 提供了一个有力的证据,证明甲基α-和β-吡喃葡萄糖甙在酸性甲醇中的异构化,是通过甲氧基的离去而给出葡糖基正离子进行的.这些化合物的水解也应该以一个类似的方式进行,先给出同样的正离子, 后被水捕捉而产生水解产物.由于只有在α-糖甙的情况下,甲氧基的离去可以在一电子对的帮助下发生,所以α-顶端异构体的水解速度应该比β-顶端异构体的快.
文献中所描述的有关缩醛水解相对速度的早期研究,没有提供有利于立体电子控制的证据.Feather 和Harris 研究了 10 对顶端异构的烷基吡喃葡萄糖甙,发现含有平键甲氧基的β-顶端异构体比带有直键甲氧基的α-顶端异构体的水解速度快(1.3—3.2 倍).通过测量 11 对烷基吡喃葡萄糖甙顶端异构体的水解速度,BeMiller 和 Doyle 观察到了类似的结果.在芳基糖甙的情况下,α-顶端异构体的水解速度略微快一些.van Eikeren 制备了构象刚性的模型化合物 125 和 126,发现直键异构体 125 水解的相对速率是 126 的 1.5倍.
Chandrasekhar 和 Kirby 观察到,在 pH7—10 的范围内,直键对硝基苯氧基异构体 127 的水解速度与 pH 值无关,并发现在相同的条件下,这一自发水解的速度比平键异构体 128 慢 3.3 倍.这样,在一个设计排除了所有其它因素(包括与酸催化反应相联系的解释问题)的体系中,没有发现缩醛断裂受立体电子控制的证据.
因而可以得出结论:对于缩醛的水解速度,电子效应不是重要的,但只有当这些化合物在它们的基态构象水解时,这才是正确的.实际情况很可能是完全不同的.例如,一个缩醛可能倾向于通过一个较高能量的构象而发生立体电子控制的水解.这样,127 和 128 的水解可能是在立体电子控制下,以一个或大或小的竞争速度发生的.化合物 127 将通过它的基态构象水解,而 128
通过船式构象 129 水解.这种情况是可能的,因为在 127 和 128 中,对硝基苯氧基团断裂的活化焓接近于 25kcal/mol,和船式构象 129 的形成能垒相比, 这是一个大得多的数值,而后者的数量级为 10kcal/mol.因此,129 能够位于反应坐标上.
缩醛水解受立体电子因素控制的第一个实验证据,是由 Kirby 和 Martin 提供的.他们测定了三环缩醛 130 和相应的顺式异构体 131 的酸催化水解
(0.1mol/L HCl)速度,130 没有电子对与离去基团处于反式共平面,而 131 却确有一处于合适取向的电子对排斥离去基团.他们发现,顺式异构体 131
迅速地被水解,而反式异构体 130 中缩醛中心的构象是刚性的,经过几周之
后,它仍没有完全被水解,这说明速度相差起码是 3000 倍.
他们也发现,在 pH9 时,131 的水解比 130 快 2.4 倍.但已证明这两个水解反应的决速步是不同的.在 130 中,决速步是 C—O 键的断裂,而在 131 中
是相应的氧鎓离子的水合.因而,在 130 和 131 的自发水解速度之间不能作直接的比较.但是,130 可以与反式双环缩醛 128 进行比较.在基态,它们基本上都有同样处于平键的离去基团,而且在 pH9 时,它们都通过决速的 C—OAr 断裂而水解.唯一的区别在于,128 可以采取船式构象 129,而 130 不能采取一个类似的构象.化合物 128 在39℃的水解速度比 130 在100℃的水解速度快20 倍,这相当于一个 104 的因子.Kirby 和 Martin 得出结论,认为这一速度差是 130 具有刚性构象的直接结果.事实上,130 不存在一个合适的构象,即不存在一个电子对与离去基团处于反式共平面的构象.因此,C—O 键断裂的立体电子能垒估计可达 7kcal/mol.
Kirby 和 Martin 发现了在缩醛断裂中,立体电子控制的一个进一步的例子,他们对直键取向的对硝基苯氧基缩醛 132 和 133 的自发水解进行了研究. 从这些化合物失去对硝基酚负离子,将产生氧鎓离子 134,它是一个带有更好的离去基团的缩醛,该离去基团就是醛的氧.如果在 134 中,A 环的氧原子上的电子对之一处于一个可参与反应的位置,那么它就应该引起一个协同反应而直接形成 135.只有在顺式异构体 133 的情况下,这种参与才是可能的. 因此,反式异构体 132 的水解速度应该慢得多.实验结果与这一预料相符合.
反式异构体 132 的自发水解比对硝基苯氧基四氢吡喃(136)慢 1380 倍,而
136 的水解比顺式异构体 133 大约快 7 倍.与反式异构体 132 中立体电子控制相联系的焓垒估计为 7.2kcal/mol,这一数值与从化合物 130 所得到的数值一致.
继 Bürgi 和 Dunitz 开拓性研究后,Jones 和 Kirby 进一步发现了键长和反应性之间的线性关系.他们首次测定了一系列 2-取代的芳氧基四氢吡喃衍生物的晶体和分子结构.在前面,我已指出,这些化合物通过一个氧鎓离子自发地水解,而且晶体结构表明,键长的差异呈现在基态里.他们发现,在直立取向的芳氧基四氢吡喃中,环内 C—O 键缩短了很多,离去基团的 C—OAr 键增长了,变化的数量依赖于 O-芳基的氧原子的电负性.他们进而注意到,这一键长变化与这些缩醛的水解速度有很简单的相关性.事实上,当 C—O 断裂是决速步时,水解速度对于离去基团的 pKa 表现出一种线性依赖关系.键长对离去基团的pKa 所作的曲线等价于键长对在水中C—O 键断裂的活化自由能所作的曲线.这曲线表明,缩醛基的两个 C—O 键的长度线性地依赖于离去基团的 pKa.按这种方式,他们可以预测一个典型的甲基吡喃葡萄糖甙的直立 C— OCH3 键长应该为 1.401Å,这一数值,在实验误差范围内,与通过 X 射线分析得到的观测平均值 1.405Å 相一致.
最近,通过研究双环羟丙基缩醛 137 的温和酸环化过程,得到了缩醛基
形成过程中存在立体电子控制的第一个实验证据(图 18).在室温下,化合物 137 只生成顺式三环缩醛 138,只有在五天之后,才出现了少量的反式三环缩醛 139.然而当顺式缩醛 138 在相同的条件下进行回流时,发生了异构化,产生了顺式缩醛 138(45%)和反式缩醛 139(55%)的一个平衡混合物.
很清楚,137 专一地转化为顺式三环缩醛 138,是一个动力学控制反应的结果.因此,反式缩醛 139 的形成与顺式缩醛 138 的形成相比,前者一定涉及
一个较高的能垒.环化反应中的第一步必定是由起始物 137 失去一个甲醇而生成环状氧鎓离子 140.然后羟基有选择地进攻氧鎓离子的β面或α面,分别生成顺式或反式三环缩 138 和 139.可容易看到,对 140 的β进攻可以在立体
电子控制下给出顺式缩醛 138,因为 138 中 B 环上的氧有一电子对与新形成的 C—O 键处于反式共平面.另一方面,对 140 的受立体电子控制的α进攻, 则不能直接形成其具有较稳定构象的反式缩醛 139.α进攻(即 141)必须首先给出反式缩醛的构象体 142,其 B 环具有船式,以便 B 环的氧原子有一电子对与新形成的 C—O 键处于反式共平面.而后 142 经过一个构象变化,成为反式缩醛的较稳定的构象 139.很清楚,转化 140→141→142→139 比转化 140
→138 需要较多的能量,这是一个和实验结果相符合的结论.
在较近期的一个研究中,使用双环硫醇半缩醛 75(图 13)获得了类似的结果.在动力学控制的条件下,75 环化生成顺式硫代缩醛 76;没有观察到反式异构体 77.这样,SH 基团对环状氧鎓离子的进攻也一定优先在立体电子控制下发生.
有些化合物中的缩醛基被固定为一种特定的构象,用这些化合物所得到的这些新近结果清楚地说明,立体电子因素对于缩醛的反应性的关系是重要的.因而,在缩醛构象不固定的化合物中,立体电子效应必定起着一种作用. 据此,下面的结论似乎非常清楚,α-糖甙一定通过它们的基态构象水解,而β-糖甙一定首先采取一个船式构象,以满足立体电子的要求.有趣的是,van Eikeren 在有关直键和平键双环异构体 125 和 126 相对水解速度的研究中, 已经得到的结果表明,两个顶端异构体经不同的过渡态水解.另外,与平键顶端异构体的过渡态相比,直键顶端异构体的过渡态包含有较广泛的 C—O 键断裂,这完全与上面有关α和β-糖甙的结论相符合,因为在顶端异构中心的 C
—OR 键断裂的过程中,当分子为椅式而不是船式存在时,过渡态中的 C—OR 比较长,而且椅式又处于较低的能量水平.
已知,在溶菌酶寡糖络合物中,从β-糖甙衍生的吡喃环扭曲了正常的椅式构象而成为半椅式构象.这一扭曲,提高了底物的基态能量,于是降低了键断裂的活化能.这样在溶菌酶催化的β-糖甙的水解中,水解经过一个扭曲的底物而发生,这主要是由于立体电子的原因.
Chwang、Kresge 和 Wiseman 做了一个有趣的观察,发现尽管烯基醚的水合通常比类似烯的水合快 105-8 倍,但桥烯基醚 143 在桥头碳上的水合比 144
慢 102 倍.还发现,含硫类似物(143,O=S)仍然比 144 的反应性差.这些值得注意的结果可以用下面的事实来解释,氧(或硫)原子上的电子对没有处于合适的排列,以给出离域的氧鎓离子.氧电子对垂直于双键的π体系,因 而143 的质子化一定产生一个高能量非离域的正离子,这个正离子进一步为氧原子的诱导效应去稳定化.于是,水合反应的总速率降低很大.
