α,β-不饱和酮
在负离子对共轭体系的 1,4-亲核加成中,立体电子效应也应该起重要的作用.因而,这些效应应该影响 Michael 反应以及α,β-不饱和酮的氢氰化反应.对这些反应的研究证明,在动力学控制下,亲核试剂对环己烯酮衍生物的加成,确实是受立体电子效应控制的.
考虑一个构象刚性的环己烯酮如 76,在立体电子效应控制下,亲核试剂从上面对它进攻,产生类船式烯醇负离子 77;而从下面进攻,则给出类椅式烯醇负离子 78.因而正如 Toromanoff 所指出的,第二个过程应该是有利的.在(一)-香芹酮(79)的共轭氢氰化反应中,Djerassi 和他的同事们
观察到,形成的主要产物是含直键氰基的差向异构体 80.与此类似, Alexander 和 Jackson 发现,底物 81 专一地生成直键氰基化合物 82 和 83.
而且在此反应条件下,两个差向异构体 82 和 83 是可以相互转化的.这些结果清楚地表明,类椅式烯醇过程 76→78 是有利的途径.
现在可以考察双环烯酮 84,其 A 环可以采取两种构象 85 和 86.在立体电子控制下,氰负离子从α面进攻构象 85,给出类椅式的中间体 87;从β面进攻构象 86,给出类椅式的中间体 88.从 85 的上面或 86 下面的趋近都不用考虑,因为这导致具有类船式构象的中间体.中间体 87 是以反式稠合的,而 88
是顺式稠合的,根据立体效应,87 比 88 稳定.因此,α-氰基加成物 89 的形
成应该比β-氰基加成物 90 占优势.这一预测得到数个实验的支持.这些实验
表明,在动力学控制的条件下,类型 84 的烯酮主要给出α-氰基加成物 89. 读者要了解详细的资料,可看 Nagata 和 Yoshi-oka 最近所写的评述文章.
有趣的是,反式烯酮 91 和 92(R=H 或 CH3)的 A 环基本上是构象刚性的.因而氰负离子从α面的加成应该比从β面的加成占优势,前者通过类椅式过渡态(93→94)发生,而后者只能通过类船式过渡态(93→95)发生.Agami, Fadlallah 和 Levisalles 最近观察到,在严格的动力学控制条件下,用反式烯酮 91 和 92,只观察到对α面进攻产生的直键氰基异构体.他们进一步观察到,在热力学控制的条件下,双环的α-和β-氰基异构体 96 和 97 是可以相互转化的,而给出一个平衡混合物.并且,正如依据立体电子效应所预测的, 直键氰基异构体 96 以快得多的速度达到平衡.
和这个双环系列相反,由甾族烯酮 92(R=H 或 CH3)衍生出来的α-和β
-氰基异构体的平衡不能发生.并且用氢氰化反应的方法制备β-氰基异构体 也是不可能的.这个结果可用下面的事实来解释,即对于甾族系列,氰基和类船式构象 95(见 98)中 C 环上的 C-11 亚甲基之间,存在着强的立体相互作用.因而在这些化合物中,从未发生过β面的加成.在氢氰化反应的实验条件下,1α-氰基胆甾烷酮(从 92 衍生得来,R=CH3)容易与标记的氰负离子(13CN)交换,这个事实证实了上述看法.
有关 Michael 反应的立体化学研究做得很少.然而 Abramo-vitch 和Struble 发现,当丙二酸二乙酯的钠化合物(从乙醇钠和乙醇中游离出来的) 加入到含 4-t-丁基-1-氰基环己烯(99)的甲苯沸腾溶液中时,化合物 101
是主要产物.这个结果可以这样解释,对 99 的直键进攻,首先给出具有类椅式构象的 100,而后经过分子内“捕捉”(看箭头)转化成 101.但是,当在热力学控制的条件下,在乙醇中进行丙二酸二乙酯负离子的加成时,得到的产物是带有平键丙二酸酯基团的 103,可能是经过扭船式中间体 102 进行的.
最近,Irie 和他的同事们观察到,丙酮二羧酸二甲酯对二烯酮 104 的两次 Michael 加成反应,给出顺式十氢合萘产物 106.
这个结果说明,中间体 105 经历了一个立体电子控制的分子内 Michael 加成而生成 106.如果在 Michael 加成中没有立体电子控制,就没有显著的理由阻止反式异构体 107 的形成.但是,如果考虑这一因素,那么对分子模型的
观察表明,得到异构体 107 看来是不可能的.
House 和 Fischer 发现,二甲基铜锂和烯酮 108 反应,产生反式和顺式 3, 5-二甲基环己酮 109 和 110 的混合物,其比例是 98∶2.Allinger 和 Riew 在有氯化亚铜存在的条件下,用甲基碘化镁与 108 反应,观察到了类似的结果. 另外,Heathcock 和他的同事们观察到由烯酮 111 专一地形成反式异构体112,而没有检测到顺式异构体.这样,铜试剂进攻的优势方式也是 76→78, 生成的烯醇负离子具有类椅式构象.
Luong-Thi 和 Rivière 发现,有机铜试剂对 4-甲基-2-环己烯酮(113) 的共轭加成,生成反式和顺式环己酮 114 和 115 的混合物,其中反式异构体
是主要的(≈9∶1).立体电子效应预测,对最稳定的构象 113 的进攻,产生类椅式的烯醇离子中间体,应该给出顺式异构体 115.因而主要形成反式异构体是出人意料的.然而可能是由于有机铜试剂和113 中C-4 上的甲基之间的强立体相互作用,抑制了顺式异构体的形成.
烯酮 113 可以采取两种不同的构象 116 和 117.从上面进攻最稳定的构象116,生成类椅式烯醇负离子 118;而从分子平面的下面进攻,产生类船式烯醇离子 119.另一方面,从下面进攻不太稳定的构象 117,给出类椅式中间体120;而从上面的进攻却产生类船式中间体 121.类船式中间体 121 的两个基团(R 和 Y)处于顺式,它的形成可容易地被排除掉.生成顺式异构体的类椅式中间体 118,必须与生成反式异构体的类船式中间体 119 和类椅式中间体120 竞争.进入的亲核试剂和 C-4 上烷基之间呈现空间阻碍的可能性,只存在于 118 的形成过程中.因此这个额外的空间因素不利于顺式异构体的形成.
Rivière 和 Tostain 的研究,为证明这确实是正确的解释,提供了令人信服的证据.他们研究了铜催化的甲基格氏试剂对 4-取代环己烯酮的共轭加成,发现当 4-烷基的大小从甲基增大到丙基时,反式和顺式之比即从 72∶28 变化到 89∶11.他们还发现,用 113 作为底物,随着试剂(RMgX,R=CH3,C2H5 和(CH3)2CH)烷基大小的增大,反式异构体的相对百分比从 72%增加到 89
%.
有趣的是,4-t- 丁基环己烯酮氢氰化反应的动力学产物不是顺式的124,而是反式的氰基酮 123.我们已经看到,有很好的证据证明,立体电子效应在共轭烯酮的氢氰化反应中起着重要作用.因此这个结果可用上面的立体证据来解释,根据这一立体效应,顺式异构体 124 的形成是不利的.
正如 Posner 在其综述中所描述的,有机铜试剂对双环烯酮如 125 的共轭加成,主要给出相应的顺式十氢合萘产物 126.我们已经知道,在这种类型的双环烯酮中,A 环可以采取两种不同的构象(见 85 和 86),在立体电子的控制下能给出一个反式产物(从 85)或者一个顺式产物(从 86).使用简单的亲核试剂,反式异构体的形成总是有利的.使用有机铜试剂,得到了相反的结果,这说明有一些其它因素,使得过渡态 86→88(CN=R)比过渡态 85→87
(CN=R)有利.
Piers 及其同事们报道,在氯化亚铜的催化下,异丙烯基溴化镁对双环烯酮 127(R=H 或 CH3)的 1,4-加成反应,专一地生成异丙烯基处于直键的
双环酮 128(R=H 或 CH3).有趣的是,由于 A/B 环以反式稠合,这些烯酮基本上是构象刚性的,它们的反应性,与根据立体电子效应所作的预测符合.
然而α,α′-二甲基八氢合萘酮 129 和三环烯酮 132 的行为与上面的不同.Marshall 和 Andersen 从 129 得到异构体 130 和 131 的混合物(当 R=异丙基时,54%的直键和 46%的平键;而当 R=甲基时,81%的直键和 17%的平键),而 Spencer 及其同事们从 132 得到 133 和 134 的混合物,比例大约为
1∶1.和具有角甲基的烯酮 127 相比,在 129 和 132 中,甲基换成了氢,这必定使得试剂从上面的进攻变得容易一些,从而有相当一部分反应可以经过类船式过渡态给出具有平键烷基的产物 131 和 134.
ouse、Respess 和 Whitesides 的报告表明,二甲基铜锂与不饱和酮 135 反应,专一地生成酮 136,其甲基和叔丁基处于反式.在这个例子中,双键处于环外,立体电子效应允许从两个面的任一面进攻.这样,136 的专一形成一定只是由于立体的原因,即平键进攻是有利的.
总之,在对α,β-不饱和酮进行 1,4-共轭加成的情况下,为了避免立体相互作用,一些底物通过一个船式构象反应,给出平键取代的产物;但当没有立体相互作用时,通过椅式构象的直键进攻在能量上是有利的.然而,这两种过程都是受立体电子控制的.
对α,β,γ,δ-二烯酮的 1,6-加成,也受立体电子效应的支配.从二烯酮 137 下面的加成,生成类椅式中间体 138;而从上面的加成,生成类船式中间体 140,才能满足轨道的最大重叠.另外 140 中的 R 基团,遭受到一个重叠的 1,2-R/H 相互作用和一个更重要的 1,4-CH3/R 立体相互作用,后者就像环己烷扭船式的船头桅杆那样.根据Marshall 和Roebke 的这一分析可预测,反式产物 139 应比顺式产物 141 占优势.这一看法得到了他们关于 1, 6-加成反应的立体化学的工作的支持.他们使用甲基、乙基、异丙基和三级丁基的格氏试剂,在乙酸铜的催化下,对双烯酮 137 进行加成,结果如下:用CH3MgI 得到 93%的直键异构体 139,用 C2H5MgBr 得到 98%139,用(CH3) 2CHMgBr 和(CH3)3CMgCl 都得到 100%139.Campbell 和 Babcock 经研究发现,在氯化亚铜的催化下,甲基碘化镁与各种甾族 4,6-二烯-3-酮反应,主要生成 C-7 甲基处于直键的 1,6-加成物.
双环双烯酮 142 用丙二酸二甲酯和叔丁醇钾,在叔丁醇中于 25℃处理 10 天,得到直键异构体 143,产率 51%,而没有分离到平键异构体 144.当反应在回流下进行时,得到 143 和 144 的混合物,其中较稳定的平键异构体是主要产物.
处于羰基β位的离去基团进行消除反应,产生α,β -不饱和体系,也应该是受立体电子控制的,即离去基团的 C—Y 键应该与烯醇离子的π体系平行
(145→146).我们从逆氢氰化反应,已经看到这一证据.
β-羟基酮在碱性条件下的脱水反应,为上述看法提供了进一步的证据. 例如,147 进行脱水,可以分别通过烯醇离子 148 和 150,产生顺式或反式的共轭烯酮 149 和 151.反式产物 151 的形成是有利的,因为在构象 150 中,平
面的烯醇负离子体系和β-碳上的苯基之间,存在较小的立体相互作用.换句话说,在导致反式产物的过渡态中,两个大的苯基是不重叠的.因而是立体效应和立体电子效应共同控制着这一反应.
有关三角形体系关环的 Baldwin 规则如下:3-至 7-Exo-Trig 过程(152
—156)都是有利的过程.3-至 5-Endo-Trig(157—159)都是不利的,但 6- 和 7-Endo-Trig(160—161)则是有利的.文献中到处是 3-至 7-Exo-Trig 过程的例子:例如,ω -羟基酸和ω-羟基酯的内酯化反应属于这一类型,从ω- 氨基酸形成内酰胺和二酯的 Dieckmann 关环反应也都属于这种类型.
Baldwin 及其同事们报道,在碱性条件下,使羟基烯酮 162 和 164(R=H 或 OCH3)环化的所有试图,都没能给出相应的呋喃酮 163 和 165(R=H 或OCH3).通过在氘代甲醇中,用甲醇钠进行尝试的环化反应,证实了这些体系对烷氧负离子亲核试剂的共轭加成的敏感性.在这些反应条件下,分离到了 162 和164(R=OCH3)的α-氘代类似物(见 166).氘原子与羰基α碳的结合,是由于甲氧负离子的可逆加成,产生一个加成物如 167,167 又经过氘交换和随后的甲醇消除.
另一方面,他们证明,在相同的碱性条件下,呋喃酮 163 和 165(R=H 或 OCH3)都迅速和充分地交换它们的α-氢原子,而在这样的条件下,162 和164 不能转化成 163 和 165.这证明不关环和不开环,是动力学阻碍的结果, 而不是热力学阻碍的结果.这样,5-Endo-Trig 过程(159)确实是一个在立体因素上不利的反应.
羟基烯酮 162 和 164(R=H 或 OCH3)的酸催化环化反应,给出相应的呋喃酮 163 和 165.这些反应的成功,归因于围绕烯酮双键旋转的能垒降低(见168A 和 168B),从而允许环化反应的发生(168B 的环化是一个被允许的 5- Exo-Trig 过程).
酚烯酮 169 用甲醇钠处理,回收到没有变化的原料.然而呋喃酮 170 在相同的碱性条件下进行处理,平稳地给出烯酮异构体 169.因而,169 不关环是一个不利平衡的结果.由于共振结构 171B,过程 170→169 可以看作是一个5-Exo-Trig 过程.
在另一个实验中,不利的 5-Endo-Trig 过程(159)处于与有利的 5- Exo-Trig 过程(154)竞争的条件下.羟基共轭烯酯 172 用各种碱处理都能有效地、单一地进行关环,给出内酯 173(5-Exo-Trig 过程),而不伴有微量的四氢呋喃 175(5-Endo-Trig 过程)产生.另外,173 能平缓地加上甲氧负离子,而产生醚 174.这表明,其双键对 Michael 类型的加成反应是非常敏感的.其次,在 172 转化成 174 的条件下,酯 175 交换它的α氢原子,但不转变
成 172.
172 的一个含氮类似物也已研究过.氨基二酯 176,从它的稳定盐酸盐中释放出来后,在 25℃通过有利的 5-Exo-Trig 路径,迅速地环化,生成内酰胺 177(100%).没有观察到产生环状氨基二酯 178 的不利的 5-Endo-Trig
过程.在另一方面,已知一级胺对α取代的丙烯酸酯进行 1,4-加成(179→ 180),比它们被酰化成α-取代的酰胺(179→181)要来得快.
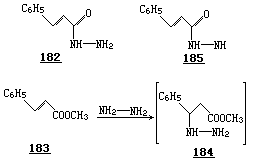
肉桂酸衍生物与肼的反应,也和上述的研究结果符合.甚至在 200℃,酰肼 182 都不能转化成吡唑啉酮 185(5-Endo-Trig 过程).但是肉桂酸酯与肼在 65℃反应,通过 1,4-加合物 184,再接着进行有利的 5-Exo-Trig 关环, 给出单一的产物 185.
与通过 5-Endo-Trig 途径关环的困难相比,6-Endo-Trig 反应是容易发生的:α,β-不饱和酮 186 在甲醇中,用甲醇钠处理,可平稳地环化成 4- 色满酮(187).
对于六员环的形成,6-Exo-Trig(155)和 6-Endo-Trig(160)过程都是有利的.然而当两种关环方式可供选择时,Exo 路径是较快的关环过程,正如 Baldwin 和 Reiss 所表明的.用各种碱(NaH,CH3ONa,t-BuOK)处理反式庚烯酸酯 188,通过 6-Exo-Trig 路径,迅速导致定量产率的四氢吡喃 189. 与此相对照,在相同的条件下,酯 190 比较慢地进行环化,生成α-亚甲基内酯 191,并且没有给出 6-Endo-Trig 方式的产物,即四氢吡喃 192.另外发现, 在 190→191 环化的条件下,192 容易交换其酯基的α质子.191 的形成相对较慢,这归因于这样的事实,即,起始物倾向于以 S-反式构象 193(而不是 S- 顺式 190)存在,这种构象的 6-Exo-Trig 关环,在空间上是不可能的.与此相反,酯 194 到内酯 195 的关环(6-Exo-Trig 过程),即使在中性条件下, 其速度也是极快的.
Baldwin 和 Kruse 发现,烯醇盐 196A—B(M=Li 或 K)给出烯醇醚 197, 而没给出环戊酮 198。和这个结果相反,在相同的条件下,烯醇盐 199A—B
(M=Li 或 K)只给出环己酮 201.在这种情况下,没有烯醇醚 200 形成.
Baldwin 得出结论,这两个环化过程之间的明显差异,产生于两可亲核试剂(即烯醇离子)烷基化反应的立体电子控制.对于这样一个离子,碳烷基化反应要求亲电试剂垂直于烯醇平面进攻,而氧烷基化反应要求在烯醇平面内进攻.结果,在五员环的情况下,C-烷基化过程 196A—198(这可认为是一个 5-Endo-Trig 过程)在立体上是困难的,但 O-烷基化过程 196B—197(5- Exo-Tet 过程)却不困难.
在最近的一篇论文中,Seebacb 和 Golinski 提出了一系列规则,用来解释在 Michael 受体 202 与 Michael 给体 203 的缩合中,苏式构型 204 的优先形成.规则如下:
-
围绕着新形成的键,所有键必须处于交叉式取向.
-
给体的 C=C 键与受体的 C=A 和 C—H 键之间,必成邻位交叉排列.
-
给体部分较小的取代基氢原子,必须处于 C=A 键的反式(反式共平面)位置.
因而,这两部分的优势趋近可由 205(或通过 Newman 投影式 206)说明. 当受体具有构型 207 时,实际的给体和受体基团依然相互靠近(参见 208). 这个拓扑规则容易解释开链硝基烯烃 209 与开链烯胺 210 的反应产物 211(> 90%立体选择性).See-bach 和 Golinski 进一步指出,有几个缩合反应也可以用这种方法来解释:(a)由烯和碳宾形成环丙烷,(b)与醛的 Wittig 反应产生顺式烯烃,(c)从亚烷基三苯基砷和醛产生反式二烷基环氧乙烷,
- 烯酮和环戊二烯的[2+2]加成,(e)E-硅基氮酸酯和醛的反应,(f)
酮、酯、酰胺和醛等的顺式和反式锂化物与相应烯醇硼化物的反应,(g)Z- 烯丙基硼烷和醛的反应,(h)E-烷基硼烷或 E-烯丙基铬衍生物和醛的反应,
(i)从环己酮形成的烯胺和肉桂醛的反应,(i)E-烯胺和 E-硝基烯烃的反应,(k)从环烷酮形成的烯胺和苯乙烯基砜的反应.
