第二章 缩醛基与相关的基团
2.1 缩醛的构象
一个缩醛基可以采取图 1 所示九种邻位交叉构象中的任意一种.构象体A、B 和 D 分别为构象体 A′、B′和 D′的镜影.剩余的几个构象体 C、E 和 F 各具有一个对称平面.因而,缩醛基原则上可以六种不同的构象 A—F 存在. 将要描述的实验结果表明,这些不同构象体的相对稳定性决定于立体电子效应和标准的立体相互作用.
早已认识到,立体电子效应影响缩醛的构型和构象,尤其在碳水化合物中,这些效应首先被发现,并用术语顶端异构效应(ano-meric effect)和外-顶端异构效应(exo-anomeric effect)进行讨论.顶端异构效应一词在1958 年为 Lemieux 所引入,用来表示吡喃环的 C1 甲氧基尽管存在着不利的立体相互作用,但仍采取直键而不是平键的倾向.外-顶端异构效应也为同一作者所引入,它所涉及的是顶端异构中心上烷氧基的 O—R 键的优势取向.
关于顶端异构效应的根源存在两个学派的想法.一派认为这一电子效应为去稳定化的因素,起因于偶极-偶极或电子对-电子对(兔耳效应)的相互作用而引起的排斥作用,可用结构 1 中的双箭头来表示.另一派认为顶端异构效应为一种稳定化的电子效应.当氧原子上的电子对与极性的 C—X(X=OR, NR2 或卤素)处于反式共平面时,这一效应即可产生.稳定化作用是通过电子对从一个杂原子部分地转移到另一个电负性原子而获得的;这种电子转移可用结构 2 中的弯箭头表明.
实际上,无论人们把顶端异构效应认为是去稳定化的因素还是稳定化的因素,都不会造成太大的差异,因为可以得出相同的关于构象体或异构体的相对稳定性的结论.例如,对于一个可采取两种构象 1 和 2 的分子,如果顶端
异构效应是稳定化因素,则得出 2 比 1 稳定的结论,因为 2 具有一个稳定化的顶端异构效应.如果某人接受顶端异构效应是去稳定化因素,那么也可得出2 比 1 稳定的结论.因为 1 具有一个去稳定化的顶端异构效应.
可能或许很可能,在缩醛基中存在着两种类型的电子效应.换句话说,2 比 1 稳定,因为相对于 1,2 通过部分电子的转移而稳定化;还因为相对于 2, 1 由于电子排斥而去稳定化.现在还没有实验技术可用来区分这两种效应.目前许多化学工作者,包括我自己,倾向于认为顶端异构效应是一种稳定化效应,而不是去稳定化效应.主要原因是,一个体系通过电子离域作用而稳定化的概念是有机化学中已充分确立了的原理,事实上,共振理论就是以这个原理为基础的.我相信,正是这一概念而不是偶极-偶极或电子对-电子对排斥的概念,使有机化学家们能更好地解释他们的结果.
顶端异构效应作为稳定化效应可用“双键-无键共振”的概念来说明,这一概念可用共振结构 4 和 3 表示,或者用等价的现代观点来表示,即这种电子离域是起因于氧原子的一个电子对轨道与 C-OR 的σ键的反键轨道的重叠.
有人也曾提出,对一个指定的醚氧原子的两个孤电子对应该进行区分, 一个较像 2p 电子对,是一个较好的电子给予体,而另一个较像 sp2 电子对, 是一个较差的电子给予体.因而,在四氢吡喃环中,氧原子的类 2p 孤电子对
不能与直键的烷氧基处于理想的反式共平面.我倾向于认为,非键的电子对是等价的,而且醚氧原子是四面体的,于是与三级胺的氮原子和四取代的碳原子类似(参见价层电子对排斥理论).然而,在缩醛基的情形中,当一个氧原子的电子对之一与 C-OR 键处于反式共平面时,如 3 中所示,另一个共振结构4 就能写出来,而真实情况应该相应于这两个共振结构的杂化体.给出电子的氧原子能与碳原子形成部分的双键,因而是部分 sp2 杂化的.通常,有机化学家并不写出所有的共振结构;他只写一个单一的结构,而在头脑中应用共振理论.我认为这是很好的实践.我也喜欢用术语“非键电子对”来代替“孤对轨道”,因为它以更准确的方式表达了化学反应性的基础(参看下文).
对顶端异构效应作出第一个精确计算,是 1968 年由 Descotes 和他的同事们实现的.这些作者研究了顺式和反式双环缩醛5 和6 的酸催化异构化反应发现,在 80℃达到平衡时,混合物中含有 57%的顺式和 43%的反式异构体. 因而,顺式异构体比反式异构体稳定 0.17kcal/mol.顺式异构体 5 有一个稳定化的顶端异构效应,而反式异构体 6 没有.在顺式缩醛 5 中,立体相互作用估计为 1.65kcal/mol(一个正丁烷的领位交叉式,0.85kcal/mol 和一个 OR 基团对环己烷成直立键,0.8kcal/mol).通过减去一个熵因素(在 80℃为0.42kcal/mol),他们得到顶端异构效应的一个数值为 1.4kcal/mol.这是由顺式缩醛 5 以两个构象(顺十氢合萘体系)的混合物存在这一事实引起的.
最近,对构象刚性的顺式和反式三环缩醛 7 和 8 的平衡进行了研究.顺式缩醛 7(平衡时占 45%)不如反式缩醛 8 稳定,能量相差 0.14kcal/mol.7 中的立体相互作用按 1.65kcal/mol 考虑,顶端异构效应就变为1.5kcal/mol,这证实了 Descotes 的数值.
首次精确估算顶端异构效应和外-顶端异构效应的数值,是通过研究 1, 7-二氧杂螺十一烷(9)而实现的(图 2).利用这一体系,通过低温核磁共振谱进行构象分析是可能的,因为每一构象变化涉及到一个具有较高能垒的椅式转化.立体效应也很容易进行估算,通过引入适当的烷基取代基,使分离以不同构象存在的异构化合物在理论上成为可能.
化合物 9 可以三种构象 9A、9B 和 9C 存在.在构象体 9A 中,两个氧原子都有一电子对与 C—O 键处于反式共平面;而在构象体 9B 中,只有一个这样的氧原子;构象体 9C 中则没有.有人假定,当在同一构象中有两个顶端异构效应时,它们是加和性的.对于一个顶端异构效应,我们接受 Descotes 的数值 1.4kcal/mol.并且只考虑电子效应,那么,9A 和 9B 应该比 9C 稳定,能量差值分别为 2.8 和 1.4kcal/mol.
然后对三种构象中的立体效应进行估算.在构象体 9A 中,两个氧处于直键.在构象体 9C 中,有两个亚甲基处于直键,而在构象体 9B 中,有一个氧和一个亚甲基处于直键.当一个亚甲基处于直键时,它相当于两个正丁烷的邻位交叉形式,每个估值为 0.9kcal/mol.当一个氧处于直键时,它相当于两个正丙基醚(O—CH2CH2CH3)的邻位交叉形式,每个估值为 0.4kcal/mol.使用这些数值,对于构象体 9A,9B 和 9C 的立体效应的估值分别为 1.6,2.6 和3.6kcal/mol.通过把稳定化的顶端异构效应和去稳定化的立体效应综合起来,构象体 9A 应该比构象体 9B 和 9C 稳定,能量差值分别为 2.4 和
4.8kcal/mol①.于是,这一分析导致如下预测:这一螺化合物一定基本上只以构象 9A 存在.这一预测已为低温下的 13C NMR 实验所证实,实验清楚地表明, 化合物 9 只以构象 9A 存在.
甲基取代的螺体系 10(图 3)也被研究过.对于这个体系,两个异构体11 和 12 都是可能的,而且分子模型表明,它们各自都可以四种不同的构象存在.对每个构象的顶端异构效应和立体效应②的估算导致如下预测,即异构体 11 只以构象 11A 存在(相对其它构象为 0kcal/mol),而异构体 12 是一个含量高的构象( 12A , 2.4kcal/mol )和一个含量低的构象( 12B , 2.9kcal/mol)的混合物.然而,由于异构体 11 和 12 是可以相互转化的(11 可以通过缩醛基的开环和再关环转化成 12 的镜影),还由于 11A 比 12A 稳定2.4kcal/mol,所以在热力学控制的条件下,应该只形成异构体 11,而且它应该只以构象 11A 存在.这一预测已完全为实验所证实.
带有两个甲基的螺化合物也被研究过(图 4).对于这一体系,有三种可能的异构体:其中的两个(15 和 16)由 dl-二羟基酮 13 经环化而成,而第三个异构体(17)从内消旋的二羟基酮 14 环化而来.在酸性条件下,15 和 16
应该容易相互转化,如果这些酸性条件足够强,并允许两个二羟基酮 13 和
- 的差向异构化,那么三个异构化合物 15、16 和 17 应该是可以相互转化的.
分子模型表明,异构体 15 和 16 均可以三种不同的构象存在;而对于异构体 17,四种不同的构象是可能的.对这些不同构象的立体效应和立体电子效应的分析表明,异构体 15 应该只以一种构象存在(15A,0kcal/mol);异
构体 16 也应该只以一种构象存在(16A,1.8kcal/mol);而异构体 17 应该以一个含量高的构象体(17A,3.1kcal/mol)和一个含量低的构象体(17B, 3.7kcal/mol)的混合物存在.
在温和的酸性条件下,dl-和 meso-二羟基酮 13 和 14 的混合物,经环化生成三种螺缩醛异构体 15、16 和 17 的混合物.低温
13C NMR 分析证实,异构体 15 和 16 具有刚性构象,它们分别以构象 15A 和 16A 存在.使用同样的技术,证明异构体 17 以构象体 17A 和 17B 的混合物存在,这与预测的一致.此外,15(或 16)的酸性平衡给出异构体 15 和 16
的混合物(≈97∶3),当异构体 17 在同样的条件下进行处理时,也转化成
- 和 16(97∶3)的混合物.这些结果与上面所做的分析完全相符.
对于三环体系 18(图 5),从二羟基酮 19 环化可能得到两个异构体 20 和 21,而且每个异构体都能以两种构象存在.但据预测,每个异构体都将具有刚性构象,分别以构象 20A(0kcal/mol)和 21A(2.4kcal/mol)存在.此外,由于异构体 20 和 21 可以通过酸性条件下的平衡相互转化,在热力学控
制的条件下,二羟基酮 19 环化生成 20 和 21 的混合物.化合物 20 证明以构象
20A 存在,当 21 在酸性条件下达到平衡时,它完全转化成较稳定的异构体 20. 也考查过带有一个甲基的三环螺体系(图 6).这一体系可以给出四种异
构体 23、24、26 和 27.异构体 23 和 24 来自于二羟基酮 22 的环化,而异构体 26 和 27 来自于异构的二羟基酮 25 的环化.在这一体系中,22 和 25 在酸性条件下不能相互转化.各个螺异构体都能以两种不同的构象存在.理论分析
① 相对稳定性用相对能量的术语来描述.
② 数值 4.0 和 3.0kcal/mol 分别用于一个甲基对甲基(或亚甲基)和氧的 1,3 二直键立体相互作用.
预测,异构体 23 作为构象体 23A(0.5kcal/mol)和 23B(0kcal/mol)的混合物存在,而异构体 24 以构象 24A(0kcal/mol)存在(图 6).因而,从二羟基酮 22 的环化反应中应该分离到大约 1∶1 的 23 和 24 的混合物.
类似的分析预测,异构体 26 应该以构象体 26A(0kcal/mol)存在,而异构体 27 应该以构象体 27A(4.8kcal/mol)存在,然而,由于异构体 26 和27 在酸性条件下是可以相互转化的,所以应该只分离到以构象 26A 存在的异构体 26.
22 和 25 的混合物在酸性条件下的环化反应,正像所预料的那样,生成
23、24 和 26 的混合物.此外,在酸性条件下,异构体 23(或 24)转化成一个大约 1∶123 和 24 的混合 T 物.而异构体 26 在酸性条件下不建立平衡.
潜伏的四羟基酮 28(图 7)在理论上可以生成异构体 29 和 30,但发现它只产生唯一的异构体 29.29 的结构已为 X 射线分析所证明.类似地,二溴二羟基酮 31 可以生成异构体 32 或 33.经过关环过程,异构体 32 是所形成的产物,而且它的结构也已被 X 射线确定.最近报道的离子载体 A-23187 的全合成证实了这些结果,它是一个聚醚抗菌剂,具有 1,7-二氧杂螺十一烷的骨架, 其构象与 29 和 32 的构象相当.
这一系列实验证实,9A(图 2)是最稳定的螺缩酮构象;它也说明在同一基团中的两个电子效应的重要性.然而,对于一个顶端异构效应来说,数值1.4kcal/mol 一定要认为是一个最小值,因为使用达到 1.7kcal/mol 这样的高数值,仍将得到类似的结论.另外,在一个取代的四氢吡喃中,立体相互作用可能比在一个简单环己烷体系中的大.由于这些原因,对于一个顶端异构效应来
说,数值 1.4kcal/mol 应该认为仅仅是一个合理的最小值.
在普通黄蜂信息素的合成中,非对映异构体 34(R=CH3)比异构体 35 优先形成,后者有一个处于平键向位的氧原子.在莫能菌素的合成过程中,Kishi和他的同事们也得到了类似的结果,莫能菌素是一种抗菌剂,在它的结构中具有 1,6-二氧杂螺癸烷骨架.有趣的是,有几个离子载体具有 1,6-二氧杂螺癸烷单元,据 X 射线分析显示,它们都以相当于 34 的相对构型存在.Descotes 及其同事们制备了几个 1,6-二氧杂螺癸烷的衍生物,并证明氧处于直键的 34(R=H)是优势的排列.他们进一步报道,化合物 36 以所示的构象存在.很有趣的是,36 中的每个醚氧原子都有一电子对与极性的 C—O 键处于反式共平面.这当然是立体电子效应的一种直接表现,这也促使四氢吡喃环以一个扭船构象存在.
1,1-二甲氧基乙烷(图 1 中,R=R′=CH3)的六种构象 A—F 现在可以给予分析了.构象体 D、E 和 F 具有两个顶端异构效应, A 和 B 有一个,而 C 没有.由于具有很强的立体相互作用,E 和 F 可以除去,并通过采用如下数值, 丁烷的邻位交叉形式为 0.9kcal/mol 和正丙醇醚(CH2CH2CH2O)的邻位交叉形式为 0.4kcal/mol,得出剩余几个构象的相对稳定性:D(0kcal/mol)>A
(1.0)>B(1.9)>C(2.9).
于是,1,1-二甲氧基乙烷应该作为一个含量高的构象体(D)和两个含量低的构象体(A 和 B)的混合物存在.对于一个烷氧基的氧,其顶端异构效应比一个四氢吡喃环氧的大,这也是可以接受的,因为烷氧基的氧原子较容易变成三角形而允许一个较大的电子离域作用.在这种情况下,构象 D 的含量
将更大.事实上,有关偶极矩的研究显示,二烷氧基缩醛以优势构象 D 存在. 最简单的缩醛,即二甲氧基甲烷,只能采取四种构象,它们对应于图 1
中的 A、C、D 和 E(R=H).构象体 E 很容易除去,其它构象体的相对稳定性估值为:D(0kcal/mol)>A(1.5)>C(2.5).这样,在二甲氧基甲烷中, 优势构象为 D.与这一结论相一致,对其气相的电子衍射研究表明,构象 D 是占绝对优势的,通过 X 射线分析,在氧亚甲基聚合物中也发现了同样的构象. 甲醛的非张力环状低聚物也是采取这种构象的.
正像我们所看到的,顶端异构效应赋予构象 D 的每个 C—O 键以一种双键的特征;因而在缩醛中,C—O 键的旋转能垒一定比在简单烷烃中所观察到的高.对于这一点,Borgen 和 Dale 可能已提供了第一个证据,他们观察到,1, 3,7,9-四氧杂环十二烷(37)的构象能垒(11kcal/mol)要比可与之相比的十二元环,如环十二烷(7.3kcal/mol)或 1,4,7,10-四氧杂环十二烷
(5.5 和 6.8kcal/mol)高得多.这也说明,37 中的二个 1,3-二氧杂基团均以与二甲氧基甲烷的构象,即 D 一致的那种构象存在.
Anet 和Yavari 通过低温质子核磁共振谱研究了氯甲基甲基醚.他们的结果表明,这个化合物以邻位交叉构象 38 存在;他们还观察到,O—CH2Cl 键的旋转能垒为 4.2kcal/mol.这一能垒明显地比只根据立体排斥作用所预测的结果高,立体能垒粗略地估计为 2kcal/mol.于是他们得出结论,顶端异构效应使 O—CH2Cl 键的旋转能垒增加大约 2kcal/mol.
st-Jacques 及其同事们最近的研究结果揭示,2,4-苯并二氧杂■呈现的环构象由取代基的本质决定,可以是椅式的 39(图 8)、扭船式的 40 或船式的 41.对于无取代基的化合物 42,观察到了椅式(39)和少量的扭船式
(40);而对于甲基衍生物 43,观察到了椅式;对于二烷基衍生物 44,45 和 46,也观察到了扭船式.
有趣的是,椅式 39 和扭船式 40 各自都有两个顶端异构效应.
在简单的 2,4-苯并二氧杂■42 中,两种形式之间的能量差可以利用椅式中 C2、C4 和 C7,上直立氢之间较大的立体相互作用来解释.在化合物 43 中,显然带有一个平键甲基的椅式 39(R1=CH3)比带有一个等斜甲基的扭船式 40
(R1=CH3)占优势,因为在后一种情形中,甲基与处于邻位交叉的亚甲基有很大的非键排斥作用.最后,在二取代衍生物 44—46 中,立体相互作用应该
使扭船式 40 比椅式 39 占有较大的优势.在这些化合物中,当它们以椅式 39
存在时,直立的烷基(R2)与 C4 和 C7 上的氢之间存在着强烈的立体排斥作用.在扭船式 40 中,部分这种立体相互作用是不存在的.
Anet 及其同事们和 Dale 及其同事们也已经证明,八元环像 1,3-二氧杂
环辛烷及一些衍生物只以扭船式构象 47 存在.构象体 47 有两个顶端异构效
应,其实质与在 40 或在二甲氧基甲烷中所能见到的一样.在上面的结果和讨论的基础上,现在就可以考虑糖甙了.对于碳水化合物、一取代的 2-烷氧基四氢吡喃和一些较刚性的体系,先前所做的努力是通过进行顶端异构中心上平键和直键异构体之间的平衡研究,估算顶端异构效应的大小.从这些研究来看,顶端异构效应的数值估计在 1.2 到 1.8kcal/mol 之间.在这些估值中,没有考虑平键和直键异构体中 OR 基团的构象;因而,忽视了外-顶端异构效应
的影响.尽管如此,这些研究还是说明了顶端异构效应的重要性.
α-和β-糖甙可以分别采取构象 A1、A2、A3 和 E1、E2、E3(图 9).这些不同构象的相对比例应该受通常的立体相互作用和立体电子效应的影响.由于立体的原因,构象体 A3 可立即排除掉.构象体 A1 有两个顶端异构效应,构象体 A2、E1 和 E2 各有一个,而构象体 E3 没有.考虑通常的立体效应和立体电子效应,这一分析预测的相对稳定性为:A1 (0kcal/mol)、A2 (1.9)、E1
(0.6)、E2(1.5)和 E3(2.5).据此,α-异构体基本上以构象体 A1 存在; 而β-异构体将是 E1 和 E2 的混合物,其中 E1 占有很大的优势.另外,在一个构象刚性的体系中,在平衡的条件下,α -异构体应该优先形成,这与实验结果相符合;Eliel 和 Giza 发现,1-甲氧基-5-甲基四氢吡喃的直键异构体比平键异构体有利,其稳定性的差值为 0.7—0.8kcal/mol.
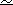 平键和直键双环缩醛
48 和 49(图 10)的酸平衡已研究过,并把其结果与单甲基缩醛 50 和 51
及同碳二甲基缩醛 52 和 53
的结果进行了比较.每对异构体基本上给出了同样的结果,即在苯中于
70℃平衡之后,有 23%的平键异构体和 77%的直键异构体(±2%).这相当于△G
0.8kcal/mol 的值,这一数值接近于对旋转异构体 A1 和 E1
的相对稳定性所做的预测
平键和直键双环缩醛
48 和 49(图 10)的酸平衡已研究过,并把其结果与单甲基缩醛 50 和 51
及同碳二甲基缩醛 52 和 53
的结果进行了比较.每对异构体基本上给出了同样的结果,即在苯中于
70℃平衡之后,有 23%的平键异构体和 77%的直键异构体(±2%).这相当于△G
0.8kcal/mol 的值,这一数值接近于对旋转异构体 A1 和 E1
的相对稳定性所做的预测
(0.6kcal/mol).
仅仅根据立体效应,平键双环缩醛 48 可以采取三种构象 54、55 和 56, 而直键异构体 49 可以构象 57 和 58 存在.平键单取代的缩醛 50 只可以采取构
象 54 和 55,而直键异构体 51 必定以唯一的构象 57 存在.最后,平键和直键
的同碳二甲基缩醛 52 和 53 基本上分别固定为构象 54 和 57.注意,54、55 和 56 对应于构象体 E1、E2 和 E3,而 57 和 58 分别对应于构象体 A1 和 A2.每对直键和平键异构体的平衡给出类似的结果.既然异构体52 和53 分别只以构象 E1(54)和 A1(57)存在,于是可以得出结论:在其它缩醛的情况下,构象体 E2(55)、E3(56)和 A2(58)的含量一定是可忽略的.已知在极性溶剂中,顶端异构效应的重要性即变小.当在甲醇中实现平衡时,就可观察到这一现象,因为直键异构体的相对百分比趋于下降(49 和 51 占 66%,53 占 62
%).
假如各种构象体有足够差异的偶合常数,构象体的相对比例就可以用核磁共振谱来确定.然而,这种方法有其不利之处,即要检测一些少量构象体是困难的.利用这种方法,Lemieux 及其同事们未能检测到β-和α-糖甙中构象体 E2、E3 和 A2 的存在.此外,也没有其它实验方法,包括 X 射线和偶极矩研究,表明α-和β-糖甙不是以 A1 和 E1 的构象存在.
构象体 E2 不能与构象体 E1 竞争,这是颇令人惊奇的,因为它们只相差一个丁烷的邻位交叉(0.9kcal/mol).实验结果表明 E1 和 E2 的能量差应该超过2kcal/mol.有人建议,这一预料之外的情况可能主要是一个短 C1—O1 键的结果.这将增大 E2 中的立体相互作用,因为一个烷氧基的顶端异构效应(外-顶端异构效应)要比四氢吡喃氧的大.通过比较糖甙中的键长(X 射线),Lemieux提出,外-顶端异构效应在事实上可能是比较强的.然而,仍然存在着这种可能性,即一小部分的 E2 与 E1 处于平衡而存在,但还有待于寻找观察这一存在的实验技术.
