其它类型的有机转化
原酸酯与格氏试剂的反应提供了一条形成缩醛和缩酮的著名合成路线(1
→2). Eliel 和 Nader 研究了这个反应的立体化学,并得出结论:它是受立体电子效应强有力地控制的.
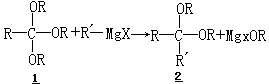
他们研究了直键和平键 2-烷氧基-1,3-二氧杂环己烷 3 和 4(R1=R2=H; R1=R2=CH3;R1=CH3,R2=H)的反应性.直键原酸酯 3 与甲基、乙基、异丙基和各种对位取代苯基的格氏试剂的反应,在室温下平稳地进行,主要生成相应的具有直键取向的 2-烷基-1,3-二氧杂环己烷 5.与此相对照,在相应的条件下,平键原酸酯 4 不能进行反应.这样,离去的烷氧基和进入的烷基都倾于取
直键位置.在化合物 3 中,二氧杂环己烷的氧都各有一个处于合适排列的电子对帮助离去基团的离去(可能受到 MgX2 或 RMgX 的催化,参见 6),而形成二氧杂环己烷的碳正离子 7.然后格氏试剂从相同的一侧进攻离子 7,产生具有直键取向的 2-烷基的反应产物.注意,格氏试剂从相反的一侧进攻二氧杂环己烷的碳正离子盐是不利的,因为它需要一个类船式过渡态(7→8).类似地, 4 中平键烷氧基的低反应性是由于这样的事实,即除非化合物 4 首先采取不利的船式构象 8(R=OCH3),否则它的离去就得不到立体电子效应的帮助.这一工作和分析发表于 1969—1970 年,是对酯基衍生的体系中的立体电子控制原理的最早贡献之一.Bailey 和 Croteau 最近报道,从 2-甲氧基-1,3-二氧杂环己烷与格氏试剂的反应形成的产物,取决于试剂-底物的配位作用和立体电子控制.
Spencer 及其同事们观察到,在温和的碱性条件下, 9、10 和 11 环化产生相应的顺-十氢合萘酮醇 13.另一方面,12 的环化生成单一的反式稠合的酮醇 14(R=H,X=H2).Spencer 得出结论,环化过程一定受固有的角取代基的大小制约:当 R 小得像氢原子时,导致反式产物(参见 15, R=H)的过渡态和导致顺式产物(参见 16 或 17,R=H)的过渡态相比,前者具有较低的能量. 然而当 R 比氢原子大时,反过来将是正确的.因此产物的形成取决于由角 R 基团和烯醇离子双键引起的立体效应.然而在这些反应中,立体电子效应仍有可能也起着重要的作用.例如,在 15 和 16 中的烯醇离子向羰基的趋近与在
17 中的不同.因而一种趋近在立体电子上可能比另一种有利,但上面描述的
实验结果未能给出这方面的信息.
表明立体电子效应在醇醛缩合中起重要作用的第一个实验结果是由Hajos 和 Parrish 报道的,他们发现,(a)三酮 18 在水中经乙酸六氢吡啶盐处理,环化为双环辛烷酮醇 19,(b)酮醇 19 用六氢吡啶处理,在 C-4 上经过一次差向异构化,产生较稳定的异构酮醇 20.这两位作者得出结论, 在动力学控制的条件下,从 18 形成酮醇 19 的结果是由于:
“⋯⋯五员环的烯醇双键和丁酮侧链中的羰基几乎是平行地排列的,而可以达到最大的π轨道重叠.”
事实上,在导致酮醇 19(参见 21)的过渡态中,烯醇双键与羰基处于反式共平面.因而这一立体化学趋近在电子上一定比导致异构酮醇 20 的有利.在后一种情况中,侧链的羰基与烯醇双键处于邻位交叉(gauche 或synclinal)(参见 22).
在 enmein 的全合成过程中, Fujita 及其同事们发现,在室温下,相应的四环酮-醛的烯醇盐 23 进行分子内的环化,只生成酮醇 24.然而同样的反应在 60℃进行时,热力学控制的条件占有优势,则得到差向异构的产物 25.
对分子模型的考察表明,动力学控制的产物 24 仍然是烯醇双键和醛双键处
于反式共平面排列的结果.同样,正像前一个例子那样,异构体 25 也得自反应基团的邻位交叉排列.
Schoemaker 和 Speckamp 报道了羟基-内酰胺 26(n=1)(HCOOH, 18 小时,室温)定量转化成螺环内酰胺酯 27(n=1)的反应.另一种可能的螺异构体 28(n=1)没有形成.在类似的条件下,羟基-内酰胺 26(n=2)也生成螺异构体 27(n=2),尽管产率较低.这些作者还报道了羟基-内酰胺 29 成功地环化成螺内酰胺 30 的反应. Evans 和 Thomas 得到了相似的结果,他们发现,烯酰胺 31 和 32 的 9∶1 的混合物在无水甲酸中环化成螺化合物 30.这个化合物在 Kishi 的 perhydrohistrionicotoxin 全合成中,是一个关键中间体.
这些结果表明,环化过程是经过一个类椅式过渡态进行的,其亚胺正离子处于假平键取向(参见 33)要比处于假直键取向(参见 34)容易.还要注意亚胺正离子和烯烃双键的相对取向,在 33 中是反式共平面的,在 34 中是邻位交叉的.
在碳环的情形中,双键的反式共平面性看来也是一个重要的因素.事实上,Harding 及其同事们报道,环己烯醇 35 的分子内环化给出螺异构体 36, 它优先于 37 而作为主要的产物(当(R=H 时,比例为 4∶1;当 R=CH4 或 n-C4H9 时,比例≥9∶1).
Schoemaker、Kruk 和 Speckamp 发现,当环的一个亚甲基被氧原子取代时,环状α-酰基亚胺离子与双键的分子内环化,采取一个完全不同的过程: 用甲酸处理 E-烯 38(在反应中产生的),给出螺双环化合物 39,而 Z-烯 40生成异构的产物 41.39 和 41 中 C11 的构型取决于起始烯的构型,因而它们的形成可以通过α-酰基亚胺离子和甲酸在双键上同步的反式共平面进攻来解
释.于是,在这个特殊的系列中,反错排列(见 42)较反式共平面排列(见43)有利,这成为 5-Exo-Trig 环化方式的第一个例子,而通常遇到的 6- Endo-Trig 环化过程却不发生.
最近, Ohloff 及其同事们进行了 dactyloxene-B(44)的八种可能的外消旋体的合成.在这个工作的过程中他们观察到,经过羟基-双烯前体 45 和 48(R1 为 CH3,R2 为 C≡CH,或相反的构型)的酸环化(p-TSA 在 CH2Cl2 中),螺双环醚 46 和 49 分别比它们的 C-5 差向异构体 47 和 50 形成得快.
已经知道,反式-二甲基前体(R1 为 CH3,R2 为 C≡CH,或相反的构型) 以构象 45A 存在,其中环的两个饱和碳上的甲基处于直立键.顺式-二甲基前体 48 采取构象 48A,这个构象在 C-2 上有一个平键甲基,在 C-1 上有一个假直键甲基.
通过考虑立体电子控制原理,可以获得 46 和 49 在动力学上优先形成的合理解释.例如,45A 的质子化应该给出烯丙位碳正离子 51.从下面对离子51 的亲核进攻(路径 A)应该比从上面的进攻(路径 B)有利,因为前者导致半椅式过渡态(52),而后者导致较不稳定的类船式过渡态(53).从 48A 优先形成 49,也可以用类似的方式来解释.
现在来考虑β-内酰胺氧负离子 54 的开环.可能有两种不同的开环方式:C—N 键断裂给出 55,或 C—C 键断裂形成 56.尽管理论计算表明,中间体 56 比 55 稳定,但在实验上只观察到了过程 54→55.
Kikuchi 提出,这个实验结果容易解释,因为过程 54→56 和过程 54→55 相比,从立体电子的观点看,前者是不利的.C—C 键断裂过程(54→56)可通过一个二阶段机理(57→58→59)来表达.在第一阶段中,产生的碳负离子
(58)具有较高的能量(没有轨道重叠),稳定化作用是通过绕 C—C 键旋转90°(→59)而获得的.这样,即使 C—C 键的断裂和 C—C 键的旋转协同地发生,在过渡态中,碳原子上得到的负电荷也没有很好地稳定化,因为负电荷和羰基π体系之间的重叠很小.
另一方面,C—N 键的断裂直接给出稳定化的酰胺离子 60.在离子 60 中, 一个氮电子对(p 轨道)通过 n-π*相互作用(初级电子效应)离域化,同时另一个电子对通过 n-σ*相互作用(次级电子效应;的确,新产生的电子对与羰基 C—Oσ键处于反式共平面)离域化.这样氮原子上的两个电子对都离域化了.
在含水四氢呋喃中,用次氯酸叔丁酯处理三环β-内酰胺 61,给出一种β-氯甲基青霉素 63.这个产率为 55%的转化可能是通过中间体氯硫化合物62 发生的.这样,在 61 中观察到 CA—S 键的特定断裂优先于 CB—S 键的断裂.与此相反,使用双环β-内酰胺 64,优先发生 CB—S 键的断裂.类似地,两个三环非对映体 65 都不发生选择性的 CA—S 键断裂.
Baldwin 和 Christie 提出,这个差异的起源几乎可以肯定是一个立体电子因素.在 64 和 65 中,CB—S 键相对于β-内酰胺的酰胺平面和 CA—S 键相对于噻唑烷的酰胺平面,CB—S 键较接近垂直于β-内酰胺的酰胺平面(参见66).因而 CB—S 键较 CA—S 键弱,并优先断裂.由于五员环的原因,使用三
环β-内酰胺 61,CA—S 键变得较接近垂直于噻唑烷的酰胺平面了(参见 67).结果键的不稳定性的顺序发生了颠倒,CA—S 键较快地断裂.立体电子控制的步骤 61→63,导致了从一个肽前体到青霉素衍生物的立体专一性合成.
Ohuchida、 Hamanaka 和 Hayashi 报道了 tromboxane A 的二硫杂类似物 71 的合成. 在关键步之一中,通过在 DMF 中使用二异丙基乙基胺
(0.2mol/L),立体选择性地实现了 3-硫基丙酸甲酯对 68 的立体电子控制的直键共轭加成.而后得到的产物 69 转化成 70,70 进一步转化成所期望的二硫代衍生物 71[(a)t-BuO-K+,HMPA,25℃;(b)NaOCH3,CH3OH;(c)0.2mol/L的 NaOH,THF].
Noyori、Kobayashi 和 Sato 报道了取代的氧杂双环辛酮 72(R=CH3,n
-C5H11,C6H5 或 CH2OR)用三氟乙酸的 Baeyer-Villiger 氧化反应.他们发现,内酯 73 和异构的内酯 74 相比,前者以一个较大的比例(大约为 2∶1 到 3∶1)产生.
内酯 73 的优先形成,可通过在相应的四面体中间体中,羟基的非键电子对的取向,并通过假设迁移的 C—C 键必须与 RCOO—O 键处于反式共平面来解释.他们还假设,过酸在 72 中羰基最小阻碍的一面进行反应.
在这样的条件下,对于四面体的中间体,只有四种不同的构象(75、76、77 和 78)是可能的.根据羟基氢和五员环之间的立体相互作用,构象 75 和
76 可以排除掉.另外构象 77 应该不如构象 78 稳定,因为前者的 R 基团和羟基氢之间存在着立体相互作用.据此,内酯 73(得自于 78)比内酯 74(得自于 77)的优先形成,应当起因于立体效应和立体电子效应的共同的作用.这个解释是基于这样一个合理的假定,即氧原子的电子对是比 O—H 键还要好的电子给体.这是为了解释实验结果,而指出羟基的立体化学(氧电子对和氢原子相对于其它键的取向)的重要性的第一个解释.
Lattes 及其同事们研究了氧杂氮杂环丙烷到酰胺(79→80)的光化学重排反应和热重排反应.他们得到确凿的证据证明,光化学过程完全是区域专一性的.他们进一步得出结论,这个过程一定受立体电子因素控制,因为在氧杂氮杂环丙烷 79 中,与氮的孤电子对处于反式的键(R″—C)比处于顺式的键
(R″—C)容易迁移.在此反应条件下(室温),在氧杂氮杂环丙烷 79 中, 通过氮反转的顺式-反式异构化作用严格地排除掉了.
他们的关键实验如下:合成了光学活性的氧杂氮杂环丙烷 81(100%的光学纯度),并确定了其绝对构型.81 的光解以 80%的产率给出光学活性的内酰胺 82(C-5 具有 S 构型),而没有观察到对映体 83.因而迁移的动力一定是与 C3—C8 键处于反式共平面的两个电子对(一个属于氮原子,另一个属于氧原子).
他们进一步指出,在氧杂氮杂环丙烷热重排中观察到的较低的区域选择性,并没有摆脱上述的立体电子要求.在这种情况下,反应所需要的能量足以导致起始氧杂氮杂环丙烷中的氮反转和 C-取代基的迁移.
Baldwin 和 Norris 报道了有关卤素(溴或氯)或环戊二烯对 1,4-二苯
醌-4-(O-甲基肟)动力学控制的加成的研究(84→86 和 84→85).上述加成反应最值得注意的结果是,具有构型 87 的产物而不是具有构型 88 的产物占有极大优势,87 中的甲氧基与保留的双键处于反式.在溴或氯加成的情形中,产生的构型 87 多于 70%,使用环戊二烯时,这种构型超过 93%.
这两个作者借助于立体电子效应解释了这些结果,这个效应涉及肟的氮上的非键电子对.他们提出,肟基的氮原子上的非键电子,对苯环上与其处于反式共平面的 C—Cσ键,产生一个小的但重要的效应.这 n-σ*相互作用使得处于反式共平面的 C—C 键具有一些反键特征,同时键长增长.这样在结构 87 和 88 中,增长的键分别是与肟的取代基成顺式的 C4—C5 和 C3—C4 键.在 88 中 C3—C4 键的增长,将减小 C-3 和 C-4 之间有效的 p-p 轨道重叠,因而减小肟氧原子上的孤对电子通过共轭体系到羰基氧上的离域化.在 87 中发生的键
增长,没有导致电子离域化作用的减小.因而这一构型比 88 稳定.在醌型衍生
物 84 中,则结构 89 比结构 90 的贡献大,这也是上述建议的一个合乎逻辑的结果.这与 Norris 和 Sternhell 为解释这些衍生物的 1H NMR 偶合常数和顺- 反平衡位置所提出的假设相符合.
Vasella 及其同事们研究了硝酮 91(R1=R2=H;R1=CH3,R2=H;R1=H,R2=CH3)对异丁烯酸甲酯的环加成,其产物为非对映异构的加合物 92 和 93,它们的差异点在 C-5 上.例如,硝酮 91(R1=R2=CH3)给出比例为 95∶5 的 92 和 93
(R1=R2=CH3)的混合物.Vasella 认为,所观察到的立体选择性是由于立体电子效应,这个效应强有力地影响着环加成反应的过渡态的立体化学.在环加成反应过程中,这一立体电子效应是由和呋喃糖环的 C—O 键处于反式共平面的氮电子对引起的(参见 94→95).当这一附加的电子离域化作用有效时,过渡态的能量即被相应地降低.
Dolson 和 Swenton 发现双缩酮 96A 和 96B(R=CH3)的酸水解,具有高度的区域专一性,分别产生单缩酮 97A 和 97B.另一方面,双缩酮 96C(R=CH3) 给出一个大约 3∶1 的 97C 和 98C 的混合物.在 96A 和 96B 水解中所观察到的选择性,归结于线性共轭的氧鎓离子 101,相对于交叉共轭的氧鎓离子 99 和100 的优先形成.另外,当趋近于转化成氧鎓离子的过渡态时,正电荷最有效的离域化要求烷氧基团处于环平面内.对于 99 和 100,这一共平面性造成立体上的拥挤;而对于 101,这种平面排列在立体上是相当合适的.对于双缩酮96C,单缩酮 97C 和 98C 混合物的形成,显然是由于在正离子 99 和 100 中, 对位甲氧基的附加稳定化作用.
Burton、Le Page、Gabe 和 Ingold 最近提出,和 4-甲氧基-2,3,5,6- 四甲基苯酚(104)相比,维生素 E(102)和相关的酚(103)之所以具有优良的抗氧化活性,是由于立体电子因素的缘故.酚的抗氧化活性取决于它们捕捉自由基的能力,正如下列反应式所示.
ROO·+ArOH→ROOH+ArO·
ROO·+ArO·→非自由基产物
 使用
4-甲氧基酚,形成的苯氧基通过未成对电子到甲氧基氧 p
轨道的离域化而进一步稳定化(105 106).在维生素 E 和化合物 103
中,这种相互
使用
4-甲氧基酚,形成的苯氧基通过未成对电子到甲氧基氧 p
轨道的离域化而进一步稳定化(105 106).在维生素 E 和化合物 103
中,这种相互
作用是允许的,但在化合物 104 中被禁止,因为前两个化合物以构象 107 存
在,而后者以构象 108 存在.在构象 108 中,由于邻位上的甲基,甲氧基发生扭曲,脱离芳香环的平面.结果甲氧基氧上电子对的离域化被禁止.
与硫相邻的碳上的氢原子趋于酸性,因而在适当的碱性条件下,用硫化物、亚砜和锍盐可以产生碳负离子.已观察到,氢原子的酸性取决于 C—H 键的立体化学.例如,Eliel 及其同事们已证明,从 1,3-二噻烷 109 衍生的平键碳负离子 110,在热力学上比直键异构体 111 稳定.在动力学控制的条件下,碳负离子 110 也优先从 109 产生,它与亲电试剂的反应是立体控制的, 产生平键产物 112(E=H 或烷基).在 1,3-氧硫杂环己烷和 1,3-二氧杂环己烷中,也证明有立体电子效应.
Wolfe 及其同事们是最早使用 D2O/OD-观察到苄基甲基亚砜中非对映异构的苄位氢之一的优先交换.然后 Baldwin 及其同事们证明,在(S)-苄基甲基亚砜(113)中,动力学上较不稳定的苄位质子的氘交换产生(R)-构型114.此后亚砜的情况广泛地进行了研究.在苄基叔丁基亚砜的 H/D 交换和烷基化中,以及在桥二芳基亚砜 115 的α-氢的交换中,发现有相当大的选择性
(速率比大于 1000).也存在有利证据证明,锍基α位的质子的酸性取决于 C
—H 键相对于硫原子上取代基的取向.
从这些结果可以得出如下结论:在这些化合物中,强的立体电子效应控制着氢的酸性.然而,这些效应的本质还需要进一步理解.一些报道表明,硫的αC—H 的酸性增强,不是由于碳负离子通过硫上 d 轨道的(d-p)π稳定化作用.这一电子效应在某些文献中曾被表述为“邻位交叉效应”.所谓邻位交叉效应,是指分子采取这样的一种结构的趋势,在这种结构中,相邻的电子对和(或)极性键之间具有最大数目的邻位交叉相互作用.Lehn 和 Wipff 进行的从头算起研究表明,由α-杂原子引起的碳负离子的稳定化作用,受到显著的立体电子效应的控制.他们的计算表明,平键型的碳负离子(见 110) 比直键型的碳负离子(见 111)稳定得多.根据这些作者的观点,这些立体电子效应可以根据 111 中的去稳定化相互作用(两个占有轨道的混合)和 110 中的稳定化相互作用(碳负离子孤电子对与反键的α*C—S 轨道混合,参见110A)来解释.Lehn、Wipff 和 Demuynck 的理论研究表明,与α-硫杂碳负离子相似,α-硒杂碳负离子也受到显著的立体电子效应的控制.
Kishi 及其同事们使用碘乙烷,观察到了申氧亚苄基二硫代-N,N′-二甲基哌嗪-2,5-二酮的区域专一的金属化(n-BuLi,THF,—78℃)和烷基化
(116→117).这个结果导致如下结论:在 116 中,H3 比 H6 的酸性强,而且这两个桥头氢的相对酸度一定取决于立体电子效应.X 射线结构测定表明 C8
—S9—C6—N6 的二面角(154°)与 C8—S7—C3—Et 的二面角(155.7°)相近.因而把区域专一性归于硫的 3d 轨道和桥头位置上的 sp3 轨道重叠的不同是不适宜的.Kishi 认为,区域专一性可能产生于硫原子孤对电子周围环境的不同.的确,S7 的两个孤电子对与 C3—C2 和 C3—N4 键的相对取向,完全不同于 S9 的两个孤电子对与 C6—C5 和 C6—N1 键的相对取向.然而,使 H3 酸性较强的这些立体电子效应的性质尚有待于进一步去理解.
硫代缩醛与两个摩尔的过氧酸的反应,通常给出差向异构的同碳二亚
 砜,差向异构体的比例受动力学和(或)平衡的控制.但是
Poje、Sikirica、Vickovi 和Bruvo
报道了同碳二硫化物系列中立体专一氧化反应的第一个例子.他们发现,使用间氯过苯甲酸可缓慢地氧化
2,2-双甲硫基-1,3-二苯基丙烷成为相应的内消旋-二亚砜.NMR
对溶液中构象性质的研究表明,内消旋- 二亚砜以构象 119 存在,这个构象和用
X 射线分析发现的在固态中的构象一样.上述作者认为,内消旋-二亚砜 119
的构型和构象是同碳二硫化物的构象
砜,差向异构体的比例受动力学和(或)平衡的控制.但是
Poje、Sikirica、Vickovi 和Bruvo
报道了同碳二硫化物系列中立体专一氧化反应的第一个例子.他们发现,使用间氯过苯甲酸可缓慢地氧化
2,2-双甲硫基-1,3-二苯基丙烷成为相应的内消旋-二亚砜.NMR
对溶液中构象性质的研究表明,内消旋- 二亚砜以构象 119 存在,这个构象和用
X 射线分析发现的在固态中的构象一样.上述作者认为,内消旋-二亚砜 119
的构型和构象是同碳二硫化物的构象
118 立体专一氧化的结果.他们进一步认为,这一特殊的立体化学过程不能用立体效应解释,它一定有一个电子的原因.
在分子轨道计算的基础上,Lehn 和 Wipff 以及 Goren-stein 和他的同事们提出:与在酯和酰胺中观察到的类似,立体电子效应在磷酸酯的水解中也起着重要的作用.例如计算表明,在三角双锥构象 120 中的直立 P—OR 键比构象 121 中的直立 P—OR 键弱,因为在前一个构象中,平键 OR 基团的氧原子有一电子对与直立 P—OR 键处于反式共平面.实验结果倾向于支持这一有趣的假设,但是在可以得出确切的结论之前,还需要进一步的实验.
