缩醛中 C—H 键的氧化
在 1971 年,发现臭氧以一种完全特定的方式与缩醛反应,生成相应的酯和醇.这一反应通过臭氧对缩醛 C—H 键的插入而进行,形成一个氢三氧化物中间体(156),这个中间体分解得到酯、醇和单线态氧几种产物.在低温下, 氢三氧化物中间体 156 可被检测到.
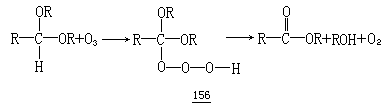
在有关这一反应普遍性的一个研究过程中,发现不同缩醛的氧化速度相差很大,这导致了如下假设,即这一反应受立体电子因素的控制.该基本的假设是:为了使氧化反应进行,要求每个氧原子上各有一非键电子对与缩醛基的 C—H 键处于反式共平面.
有关一系列适当化学模型的反应性的一个研究证实了这一假设.缩醛基的构象体 A、C 和 F(图 1)在每个氧原子上都有一电子对与 C—H 键处于反式共平面,构象体 B 和 D 都只有一个氧带有合适取向的电子对,而构象体 E 没有.这样,构象体 B、D 和 E 应该是惰性的,而构象体 A、C 和 F 对臭氧则应该是活泼的.
发现构象刚性的β-糖甙对臭氧的反应性是活泼的.β-糖甙的旋转异构体 E1、E2 和 E3(图 9)分别相应于缩醛基的构象体 A、B 和 C,因此至少其一应该是活泼的.化合物 157 和 158 是旋转异构体 E1 和 E3 的刚性模型化合物. 这两个化合物都能与臭氧反应,这样,构象体 A 和 C 都是活泼的构象体.另一方面,不活泼的化合物 159 是旋转异构体 E2 的一个刚性模型,这样,正像所预料的,构象体 B 是不活泼的.
构象刚性的α-糖甙对臭氧的反应是惰性的.α-糖甙的旋转异构体 A1、A2 和 A3 分别对应于缩醛基的构象体 D、B'和 E,因而正像所预料的,它们都是不活泼的。化合物 160 可以被认为是α-糖甙的最稳定旋转异构体 A1(构象体 D)的一个刚性模型,发现它是不活泼的.最后,作为构象体 F 刚性模型的1,3-二氧杂环己烷 161 可平稳地被氧化.
这一分析得到下面事实的支持,即发现构象易变的α-吡喃糖甙可与臭氧反应.当α和β-呋喃糖甙不保持在一种刚性构象时,也观察到了类似的结果.有趣的是,非环(二烷氧基)缩醛与臭氧的反应速度比环状缩醛如 1,
- 二氧杂环己烷 161 慢得多.一般认为,非环缩醛氧化的慢速度是由于它们以构象 D(图 1)存在的事实,这个构象的反应性是不活泼的.这样,非环缩醛在与臭氧反应之前,首先经过一个从构象体 D 到构象体 A、C 或 F 的构象变化. 大部分非环缩醛以不活泼的构象 D 的形式存在,这个事实使反应物的有效浓度降低了,因而氧化反应的速度受到了影响.另外,环状缩醛的构象体 F 可能处在一个较高的能量水平,于是它比非环缩醛能够采取的构象 A 或 C 活泼.
Taillefer 及其同事们进行环状和非环缩醛的一个有趣的动力学研究. 他们发现,环状和非环状缩醛的活化焓是非常低的(大约 5—7kcal/mol),
而活化熵却很高,而且为负值,这表明了一个高度有序的过渡态.他们也证明了两种不同类型的缩醛对臭氧的行为是不同的.对于非环状缩醛,他们观察到了一个等动力学关系.而且,发现等动力学温度低于实验温度范围,在此范围内,反应性受熵因素的控制.这些结果和对于环状缩醛所得到的结果形成对照,后者的等动力学温度高于工作温度,在这一温度范围内,反应性主要决定于焓因素.这些结果和上面的解释构成了相当有力的实验证据,证明在非环状缩醛的情况下,在氧化反应之前必须发生一个构象变化.
从上面的结果很清楚地看到,缩醛基的氧化过程受立体电子因素的控制.下一步是试图理解这些电子效应是怎样发挥作用、从而降低氧化反应能垒的.得到的实验证据表明,在氧化这一步的过程中,缩醛基的中心碳变为带
正电荷的.另外,已经测量到一个相当高的初级同位素效应(KH/KD=6.5).这些结果表明,反应机制是经过一个直接的负氢转移进行的,产生一个二烷氧基碳正离子和一个氢三氧负离子,它们结合为氢三氧化物中间体(162→163
→165)(图 20),或者是经过臭氧以 1,3-方式对缩醛基 C—H 键的插入(164
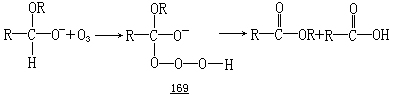
→165).如果情况是这样的话,则对于缩醛基的每个氧原子都要求各有一电子对与 C—H 键处于反式共平面,就变得很清楚了.反式共平面的电子对应该增加 C—H 键的电子密度,因而,这个键将更容易受到亲电试剂如臭氧的进攻.Lehn、Wipff 和 Bürgi 所进行的从头算起研究与这一结论相符合.如果有初生态的二烷氧基正碳离子 163 形成的话,则这个效应也将对这种正离子起稳定化的作用.这一解释与这样的事实相符,即带负电荷的半缩醛氧负离子168 与臭氧的反应比二烷氧基缩醛的快.反应产物是相应的羧酸和酯的混合
物,据推测,它们来自于中间体 169 的两种可能的断裂方式.
氢三氧化物中间体的形成也有可能是经过自由基机制发生的(166→167
→165).这意味着,当相邻氧上有两个电子对与自由基处于反式共平面时, 自由基会更稳定.文献中有证据表明,实际情况确实如此.Bernasconi 和Descotes 已经观察到,顺式和反式 2,6-二甲氧基四氢吡喃在二苯甲酮存在下的光解反应倾向于夺取直键氢;顺式异构体 170 的光降解比反式异构体171 快.Hay-day 和 Mckelvey 发现,在室温下,三线态的二苯甲酮从顺-2-甲氧基
-6-甲基四氢吡喃(172)夺取直键氢,比它从反式异构体 173 夺取平键氢大
约快 8 倍.既然两个化合物给出同样的产物分布,那么就可以得出形成了一个共同的自由基的结论.随后,通过利用 EPR 谱鉴定,从构象刚性的顺式和反式前体产生的 2-烷氧基四氢吡喃-2-自由基证实了这一结论.在室温下,使用两对构象刚性的顶端异构体-2-甲氧基-1,3-二氧杂环己烷和 2-甲基-1,3-二氧杂环己烷,也证明了叔丁氧基自由基对于夺取顶端异构碳的直键氢比夺取平键氢显示出较强的(12 倍)优势.
最近,在有关氢原子(这些氢原子是在各种环状和非环状醚,缩醛和原甲酸酯上的)被光引发的叔丁氧基自由基夺取的相对速度的一个研究中, Malatesta 和 Ingold 观察到一个显著的立体电子效应,当这些 C—H 键和具
有合适电子对取向的氧相邻时,它能高速度地从这些 C—H 键夺取氢.而当 C
—H 键和带有不合适取向电子对的氧相邻时,夺取氢的反应则慢得多.使用取代的 1,3-二氧杂环己烷,Beckwith 和 Easton 观察到了类似的结果.通过自由基的夺取反应,缩醛向内酯的转化也已有报道,在夺取反应的键断裂中存在着立体电子控制.Remy、Cottier 和 Desco-tes 已报道了α和β异构体174和 175 的光解.使用β-顶端异构体 174,反应在 40 小时内完成,产生螺化合物 176.使用α-顶端异构体 175,反应很慢,没有导致某一特定产物的形成.用各种试剂如 N-溴代琥珀酰亚胺,三乙基氧鎓盐或三苯甲基四氟硼酸
酯,把缩醛基氧化成酯也已有报道.但还没有证实这些反应是否受立体电子控制.Angyal 及其同事们用三氧化铬在乙酸中氧化碳水化合物得到的缩醛,他们在研究过程中进行了一个有趣的观察.他们发现,α和β-糖甙与这一试剂进行不同的反应.使用β-糖甙,顶端异构的氢首先被氧化,所得到的产物是酮酯(177→178),而使用α-糖甙,甲基被缓慢地氧化成甲酸酯(179→180). 在这两个反应中,立体电子效应一定又起了作用.
