第三章 酯基与相关的基团
- 立体电子效应和酯基
这一章将叙述在酯的形成和水解过程中,立体电子效应对四面体中间体的断裂的控制.立体电子效应对酯基本身的结构也起作用,所以我们首先对酯基进行讨论.
已经提出酯基中存在两种类型的电子效应:初级电子效应和次级电子效应.这两种类型的电子效应的作用和相对重要性,通过分析这两种效应与 Z 式和 E 式的几何形状及相对稳定性的关系,是可以理解的.
众所周知,酯具有平面结构,Z 式较 E 式稳定得多(约 3kcal/mol).在小环内酯中,当然只观察到 E 式,但只要内酯环的大小允许 Z 式的存在,就再也观察不到 E 式的存在.甚至在甲酸叔丁酯中,Z 式中羰基氧原子和叔丁基之间必定存在着强的立体排斥作用,Z 式仍然占有优势(~90%).
初级电子效应起因于醚氧和酯羰基之间的电子对离域作用,如共振结构1、2 和 3 所示.共振结构 1 和 2 表明了电子对在羰基碳和氧之间的离域
(12),共振结构 2 和 3 表明了醚氧有一对电子离域到同一中心碳上(23).因而初级电子效应可以看作是两个n-π*相互作用的结果.所涉及到的三个原子都可认为是 sp2 杂化的,在此基础上,三维图象 4 和 5 分别对应于酯基的 Z 式和 E 式.
酯基中的次级电子效应和前面讨论过的缩醛的顶端异构效应基本上相似,涉及到一个 n-σ*相互作用.唯一的区别在于,酯中的中心碳是三角形的
(sp2 杂化的),而缩醛中是四面体型的(sp3 杂化的).
在 Z(4)酯和 E(5)酯中,羰基氧都有一电子对与 C—OR 键处于反式共平面,因而应该存在一个 n-σ*相互作用,因为这一电子对轨道可以与 C—OR 键的反键轨道(σ*)重叠.换句话说,这个次级电子效应(或顶端异
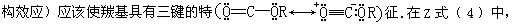
可能存在另一个次级电子效应,因为在这种形式中,醚氧有一电子对与羰基的 C—Oσ键处于反式共平面.因而,这一电子对轨道能够与羰基σ键的反键轨道(σ*)重叠.这样在 Z 酯中,除了初级电子效应外,还有两个次级电子效应,这可用三维图象 6 和二维图象 7 来说明.而在 E 酯中,除了初级电子效应外,只有一个次级电子效应,如图象 8 和 9 所示.
在 Z 式酯中,多出的一个稳定化的次级电子效应,可能比在缩醛中所观察到的(1.4kcal/mol)大,因为酯中的羰基键的极化程度较缩醛的 C—OR 键高,从而羰基σ键的反键轨道应该具有较低的能量,也就允许更大的重叠. 由此认为,Z 式酯比 E 式酯的较大稳定性(约 3kcal/mol)主要起因于这个次级电子效应.
这样初级电子效应(n-π*相互作用)形成了酯基的共轭体系,而次级电子效应(n-σ*相互作用)是非键电子对与酯基的σC—O 键处于反式共平面的结果.很清楚,初级电子效应在能量上比次级电子效应来得重要,用初级和次级电子效应这样的术语来叙述也是合理的,因为这两种效应起源于同一个
化学原理,即电子对在空间中的取向引起电子离域化作用.
除了 E 式和 Z 式的相对稳定性以外,没有其它直接的实验证据来证明次级电子效应在酯基中的重要性.然而,通过考虑这些电子效应,可以解释不同形式的二烷氧基碳正离子的相对稳定性.因为二烷氧基碳正离子是酯烷基化的衍生物,所得结果可用作支持次级电子效应在酯中重要性的证据.从 X 射线的证据得知,二烷氧基碳正离子和酯一样具有平面结构,理论上可以三种不同的形式存在,ZZ(10)、 EZ(11)和 EE(12),这些结果还得到了理论计算的支持.10 的两个氧各有一非键电子对与一个极性C—O 键处于反式共平面,11 只有一个,而 12 则没有.这样,10、11 和 12 分别有 2、1、0 个次级电子效应,在此基础上,它们的相对稳定性也应该按着这样的次序排列.而在ZZ 式(10)中,两个 R 基团存在着很强的立体排斥作用;这样,除非两个 R 都是某环上的一部分,否则这种形式一定要排除.因此,EZ 式(11)必定代表着二烷基碳正离子的最稳定形式.
Ramsey 和 Toft 通过核磁共振谱提供的第一个证据表明,由乙酸甲酯衍生的二甲氧基碳正离子只以 EZ(11)式存在.后来发现,在—30——80℃, EZ 式占有优势;而在 0—38℃,EZ 和 EE 两种形式都存在, EE 式(12)的相对百分数取决于取代基的性质.当 R1=H、R2=烷基时,EZ 式成为唯一的形式. 质子化的酯和酸也主要以 EZ 式存在.
最近,已经得到了进一步的证据说明次级电子效应的重要性,这些证据来自于对碘化物与内酯盐的 SN2 反应的研究.发现碘离子迅速与内酯盐如 13 反应,给出碘代酯 14、内酯 15 及碘代烷的混合物.例如,当 13 中 R=CH3 时, 得到了碘代酯 14(R=CH3)(70%)和δ-戊酸内酯 15(30%)的混合物.
这些实验结果清楚地表明,碘代酯的形成(过程 A)可以有效地与内酯和碘代烷的形成(过程 B)相竞争,这是一个意外的结果.在过程 B 中,形成了两个分子,环没有打开;而在过程 A 中,只形成了一个分子,环却被打开了.在此基础上,过程 A 比过程 B 应该需要较多的能量.男外,在内酯盐中有一个甲氧基,碘在一级甲基上的取代应该比在环的二级亚甲基上的取代来得容易.这个因素也有利于内酯的形成,然而主要观察到了碘代酯.因而,很清楚,一定有一种只在过程 A 中起作用的新因素,降低了其过渡态的能量,以致过程 A 能够与过程 B 相竞争.
假设这些内酯盐以 EZ 式(11)存在,产生 14 和 15 的两个过渡态可以分
别用 16 和 17 来说明.在 17 中,被断裂的键与非极性的 C1—C2 键处于反式共平面;而在 16 中,被断裂的键与极性 C1—O 处于反式共平面.换句话说,在16 中,C5—O5 键的电子对轨道可以通过与极性 C1—O1 键的反键轨道(σ*) 相互作用而发生离域;在 17 中,R—O1 键的电子对轨道不能离域到一个极性C—O 键的反键轨道上去.这样,前一过程在电子上应该比后一过程有利,据此,对实验结果的解释就变得清楚了.因此,这些结果可以认为是对酯基中次级电子效应重要性的进一步支持.
既然已经详细地说明了酯基中的电子效应,我们就可以应用水解反应的立体电子控制理论,来考察从酯得来的四面体中间体的形成和断裂,这一理论是在 1972 年提出来的.根据这一理论,为了有利于反应,对酯基的亲核进
攻必须垂直于(大约 109°,根据文献共轭体系所在的平面,并生成四面体中间体,其中两个氧原子各有一孤电子对与新生成的键处于反式共平面.遵循这条规则,一个 Z 式酯(4)与一个亲核试剂 Y 反应必定给出中间体 18(图 1);而 E 式酯(5)一定给出中间体 19.对过程 4→18 的考察表明, Z 式酯的构象转移到四面体中间体中,由于在 4 中,C—R 键和 O—R 键是处于反式共平面的,所以在 18 中还仍然处于反式共平面.与此类似,在过程 5→19 中,在 E 式酯中处于顺式的两个 R 基团在中间体 19 中将采取邻位交叉取向.同时,4
→18 和 5→19 的转变都遵从最小移动原理.
根据微观可逆性原理,其逆过程一定经过同样的途径.事实上,立体电子理论是首先通过考察四面体中间体的断裂这一过程才建立起来的.很明确,四面体中间体的构象一定要转移到反应产物中去,只有当四面体中间体的两个氧原子各有一电子对与离去基团处于反式共平面时,C—Y 键才能在立体电子的控制下发生断裂.
中间体 18 和 19 代表着四面体中间体的三种可能的邻位交叉构象体中的
两种.因而,分析第三种邻位交叉构象体 20 是恰当的.在立体电子控制下,中间体 20 不能发生断裂,因为 OR 氧没有处于合适取向的电子对来排斥离去基团,20 中离去基团的离去只可能在一电子对(来自于 O-)的帮助下发生,但由此却产生了一个非共轭的“酯”21(无初级电子离域作用),它相应于 E 式和 Z 式酯在热异构化过程中产生的高能中间体.因为这种转化的能垒至少是 15kcal/mol,所以,21 和 4(或 5)之间的能量差一定具有这个数量级. 据此,断裂过程 20→21 的能量显然比断裂过程 18→4 或 19→5 的高得多①.
中间体 18 给出酯的最稳定形式(Z),而 19 给出最不稳定的形式(E). 随之产生的一个有趣的问题是,是否 Z 酯形成(18→4)的能垒也比 E 酯形成
(19→5)的能垒低呢?分析一下次级电子效应就能够获得答案.
值得注意的是,在两个过程中,四面体中间体的电子效应都完全传递到酯基中去了.在中间体 18 中,除了与离去基团处于反式共平面的两个电子对外,每个氧都有一电子对与 C—O 键处于反式共平面.这样,18 就有四个次级电子效应(n-σ*).然而,在 18 断裂生成 Z 式酯(4)的过程中,有两个次级电子效应变成了初级电子效应(n-π*相互作用形成π体系),而另外两个次级电子效应仍然保留着(n-π*).在中间体 19 中,除了与离去基团处于反式共平面的两个电子对外,带有负电荷的氧也有一个电子对与 C—OR 基团处于反式共平面.这样,19 有三个次级电子效应,它断裂生成 5 时,其中的两个次级电子效应(n-σ*)变成了初级电子效应(n-π* ),而第三个次级电子效应保留着(n-σ*).
在每一种断裂中,除了有两个次级电子效应(n-σ*)转变为初级电子效应外(n-π*),没有任何电子效应的得失,据此,能垒应该是相似的.然而, 因为中间体 18 与 19 相比,有一个额外的次级电子效应,所以 18 的基态能量应该比 19 的低.于是,在四面体中间体中是以 18、19 和 20 构象体的混合物存在的,并通过旋转迅速达成平衡,这对 Z 式酯(4)的形成是有利的.结果, 在这些条件下,不但构象体 20,就连构象体 19,都不能与产生 Z 式酯(4)
① 在 20 中,Y 基团离去的能垒也一定比相应的半缩醛(20,OR=R,Y=OR)高,因为 20 中,OR 基团的氧的诱导效应使中心碳产生了部分正电荷.
的构象体 18 相竞争.如果确实如此的话,就可以说中间体的断裂是在初级和次级立体电子控制下发生的.
也有一些情况,不必发生构象变化就能产生相互竞争的两种可能断裂. 在一个四面体中间体中,当离去基因 Y 是另一个—OR 基时,这种情况也是可能的.例如,我们可以考虑三个四面体中间体 22、23 和 24.
在中间体 22 中,O2 的 R 基团是这样取向的,以致 C—O3 键断裂的结果将产生 Z 式酯.另一方面,因为 O3 的 R 基团和 C—O2 键处于反式共平面,所以 C
—O2 键的断裂不能发生.因而可以预测,22 将通过 C—O3R 键的断裂给出一个Z 式酯.在中间体 23 中,O3 的 R 基因处于这样的方向,可通过 C—O2 键的断裂给出一个 E 式酯.因为 R—O2 键与 C—O3 键处于反式共平面,C—O3 键的断裂不能发生.这样,23 应该通过 O2R 基团的离去给出一个 E 式酯.
最后一个例子,即中间体 24 是比较有意思的,因为它可以给出 Z 式酯, 或者给出 E 式酯.事实上,C-O3 键的断裂能够产生 Z 式酯,而 C—O2 键的断裂将产生 E 式酯.既然在同一构象体中两种断裂是可能的,前面所用的论据,即不同构象体的相对稳定性,就不能用来对这种情况进行预测了.然而,仍然可以预测在 24 中 C—O3 键的断裂比 C—O2 键的断裂有利,因为前者导致一个具有两个次级电子效应的产物(Z 酯),而后者导致一个只具有一个次级电子效应的产物(E 酯).结果,多出的次级电子效应(n-σ*)降低了 Z 酯产物的基态能量,它也必定对降低形成 Z 式酯的相应过渡态的能量起作用.
在前面有关缩醛的一章里,我们已经将顶端异构效应(或次级电子效应) 作为 n-σ*相互作用以及它们对 OR 基团离去能力的影响进行了讨论,把这个原理应用于中间体 22、23 和 24,得出了下面的结果.在中间体 22 中,O3 给出一个次级电子效应(与 C—O1),O2 给出两个(一个与 C—O1,一个与 C— O3)次级电子效应.结果,C—O3 键有一个部分双键特征,而 C—O2 键有两个部分双键特征;这样,O3R 是一个比 O2R 好的离去基团.因此,这是另一个对中间体 22 中 O3R 优先离去的有力证据.在中间体 23 中,02 和 03 各有一个次级电子效应,O3R 和 O2R 基团一定具有相近的离去能力.在中间体 24 中,O3 有一个次级电子效应,O2 有两个.这样,O3R 基团是一个较好的离去基团,它的离去应该比 O2R 的离去容易.这个因素也有利于解释从中间体 24 生成 Z 式酯优先于 E 式酯.
在更普遍的基础上,一个具有两种不同烷氧基的半原酸酯中间体如 25, 可以产生两种不同的酯 26 和 27,每种酯都具有或 Z
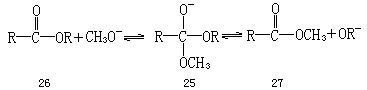
式或 E 式的构象.从理论上讲,中间体 25 可以采取图 2 中所描述的九种不同的邻位交叉构象,对每种构象受立体电子控制的断裂的预测结果列于表1.构象体 A(或 F)与中间体 22 的构象相同.类似地,构象体 E(或 G)相应于 23,构象体 B(或 I)相应于 24.构象体 D 不能断裂;因而预测,它断裂的
能垒一定比其它情况下的高.构象体 A,B 和 C 代表着甲氧负离子对 Z 式酯进攻而产生的三种可能的构象体.与此类似,构象体 G,H 和 I 是甲氧负离子与E 酯反应所形成的三种可能的构象体.
对四面体中间体断裂过程中的初级电子效应(n-π*相互作用)和次级电子效应(n-σ*相互作用)之间的相互关系,我们已经作了详细的解释,在试图解释那些能够有力地支持立体电子原理在水解反应中具有重要性的实验之前,我们必须考虑四面体中间体的断裂能垒和构象变化能垒的相互关系.事实上在某些情况下,四面体中间体断裂之前可能发生构象变化.因为中间体断裂的能垒随离子状态而变化,而离子状态又是 pH 的函数,所以必须考虑四面体中间体的各种离子状态.
